
1 Введение
Хроматография – физико-химический метод разделения компонентов анализируемой смеси, основанный на разности коэффициентов их распределения между двумя фазами: неподвижной и подвижной.
В газовой хроматографии в качестве подвижной фазы используется газ, называемый газом-носителем. Неподвижная фаза может быть, как твердым телом (адсорбентом), так и жидкостью (в виде пленки, нанесенной на поверхность твердого носителя).
Специфические понятия в хроматографии
Подвижная фаза – элюент, твердый носитель, покрытый пленкой; поток жидкости, флюида или газа, перемещающий компоненты разделяемой смеси вдоль неподвижной фазы.
Растворенное вещество, покидающее жидкую фазу вместе с элюентом, называется элюатом.
Процесс перемещения образца с элюентом называется элюированием.
Подвижная фаза может быть либо газом, либо жидкостью. Мы будем рассматривать только газовую хроматографию, главными особенностями которой являются высокие коэффициенты диффузии и использование газа с малой плотностью и малой вязкостью. Почти во всех случаях предполагается, что газ ведет себя как идеальный.
Неподвижная фаза может быть либо твердым телом, тогда основным процессом для установления равновесия, является процесс адсорбции. Если неподвижная фаза – жидкость, то, дабы избежать конвективного перемешивания и обеспечить быстрый массообмен между двумя фазами, неподвижную жидкую фазу наносят на твердый носитель. Этот носитель должен быть инертным по отношению к компонентам анализируемой смеси, чтобы не влиять на установление равновесия между подвижной и неподвижной фазами (в противном случае будет наблюдаться искажение пиков, сильное размывание хроматографической зоны, уменьшение как высоты, так и площади пика, либо нужный пик совсем не пропишется на хроматограмме).
Адсорбент – твердый сорбент, концентрирующий на своей поверхности газы, пары или растворенные вещества.
Абсорбент – твердый или жидкий сорбент, растворяющий в своем объеме газы, пары или компоненты жидких смесей.
Сорбция – процесс поглощения газов, паров или жидкостей поглотителями (сорбентами), обратный процесс называется десорбцией.
Сорбцию можно осуществить двояко:
статическая сорбция – процесс протекает при относительном покое обеих фаз и завершается установлением равновесного распределения веществ между фазами.
динамическая сорбция – процесс, в котором происходит направленное перемещение подвижной фазы относительно неподвижной
Сорбат – анализируемая смесь веществ, переведенная в жидкое или газообразное состояние.
Массопередача – явление переноса вещества в пределах одной или нескольких фаз, лежащее в основе процесса разделения разнообразных смесей.
Благодаря возможности объединения процесса высокоселективного разделения с последующим высокочувствительным детектированием, хроматография стала самым распространенным методом анализа сложных смесей, позволяющих определять до 1000 веществ в одной пробе с пределом обнаружения на нонаграммовом и фемтограммовом уровнях. В современной аналитической химии 75-80% всех анализов выполняется хроматографическими методами.
В зависимости от агрегатного состояния подвижной фазы хроматографию подразделяют на газовую (подвижная фаза – газ), жидкостную (подвижная фаза – жидкость) и сверхкритическую флюидную (подвижная фаза – флюид).
Термин "Газовая хроматография" объединяет все методические варианты, в которых подвижная фаза – газ или пар.
С помощью газовой хроматографии можно выполнять качественное или количественное определение компонентов смесей любых органических и неорганических газов, жидкостей, твердых тел, давление пара которых при температуре кипения находится в диапазоне 0,133-133 Па (0,01-1 мм. рт.ст.), то есть перегоняющихся без разложения в области температур до 4000С-5000С, или более высококипящих соединений, для которых, однако, отработана методика воспроизведения термического разложения. Могут быть анализированы даже те соединения, которые могут быть превращены в летучие производные для последующего газохроматографического анализа.
Времена выхода компонентов, отсчитываемые от момента ввода пробы до момента регистрации вершины пика, или, иначе, объемы подвижной фазы, затраченные на перенос через колонку каждого компонента, дают КАЧЕСТВЕННУЮ характеристику веществ. Сопоставление площадей (высот) пиков позволяет выполнять КОЛИЧЕСТВЕННЫЕ определения.
Кроме качественного или количественного определения, также возможно проводить препаративное выделение и очистку любого содержащегося в анализируемом образце вещества, поскольку имеется возможность осуществить полное разделение всех компонентов смеси.
2 Состав хроматографа
Газовый аналитический хроматограф – совокупность взаимодействующих систем, предназначенных для проведения анализа в оптимальном режиме хроматографического разделения исследуемой смеси с целью определения ее состава.
Устройство газового хроматографа определяется следующим понятием:
Хроматография есть процесс разделения, происходящий в результате дифференциального элюирования компонентов смеси, испытывающих равновесия распределения или адсорбции между неподвижной фазой и подвижной фазой, которая продвигается через неподвижную фазу".
Состав газового хроматографа:
Система подготовки газов (установка, стабилизация и очистка потоков газа), систему подачи в колонку газа-носителя с постоянной объемной скоростью. Эта система включает в себя автоматические регуляторы давления и(или) расхода газа-носителя.
Система ввода пробы – дозирующее устройство, позволяющее вводить в поток газа-носителя непосредственно перед колонкой определенное количество анализируемой смеси в парообразном состоянии.
Система термостатирования (температурные режимы колонок, детекторов, дозирующих устройств) помещены в термостаты, управляемые терморегулятором. Если необходимо повышать температуру кипения в процессе анализа, используют программатор температуры колонки. Терморегулятор с программатором составляет систему термостатирования, в которую может также входить устройство для измерения температуры.
В колонке осуществляется разделение смеси на отдельные составляющие компоненты.
Компоненты в смеси с газом-носителем подаются в детектор, который преобразует возникающее изменение физических или физико-химических свойств бинарных смесей компонент – газ-носитель (по сравнению с чистым газом-носителем) в электрический сигнал. Величина сигнала зависит как от природы компонента, так и от содержания его в анализируемой смеси. Сигнал детектора, преобразованный усилителем, записывается в виде хроматограммы. С некоторых детекторов сигнал может быть записан без предварительного усиления.
Все функциональные системы хроматографа взаимосвязаны, поэтому работа прибора может быть удовлетворительной лишь при условии четкой и правильной работы каждой системы в отдельности.
Рисунок 2.1 – Принципиальная схема хроматографа
Прохождение в детекторе газа-носителя без пробы на хроматограмме отражается фоновым сигналом детектора, который называется нулевой линией. Нулевая линия имеет высокочастотные колебания – шум. Изменение сигнала нулевой линии детектора во времени называется дрейфом.
При прохождении через детектор анализируемого компонента происходит отклонение уровня сигнала детектора от нулевой линии. Это отклонение отображается на хроматограмме в виде пика. Пик на хроматограмме имеет следующие характеристики:
Время удерживания. Время от начала анализа до выхода максимума пика. Время удерживания – качественная характеристика анализируемого компонента, площадь и высота – количественные характеристики.
Площадь. Область, ограниченная профилем пика и базовой линией.
Высота. Расстояние от вершины пика до базовой линии.
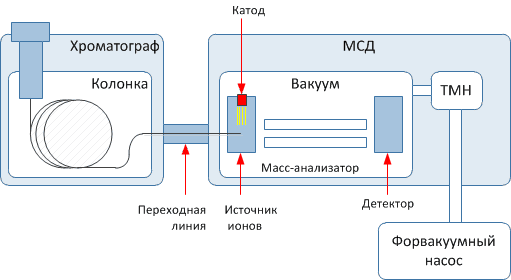
Рисунок 2.2 – Структура хроматограммы
После получения хроматограммы проводится ее обработка, которая включает в себя несколько последовательных этапов.
Фильтрация шумов. Сглаживание нулевой линии хроматограммы с целью повышения стабильности автоматической обработки хроматограммы. Фильтрация шумов обычно применяется в исключительных случаях, например, при наличии выбросов на хроматограмме. В большинстве случаев в данной операции нет необходимости.
Интегрирование пиков. Определение базовой линии пиков и измерение параметров пиков (время удерживания, площадь, высота).
Идентификация пиков. Отнесение пиков на хроматограмме к тому или иному компоненту в таблице по параметрам удерживания.
Градуировка. Анализы проб с известным содержанием анализируемых компонентов с целью вычисления коэффициентов чувствительности детектора к этим компонентам. В тех случаях, когда градуировочные коэффициенты известны заранее, градуировка не проводится. При этом известные коэффициенты заносятся в таблицу компонентов.
Количественный расчёт. Завершающая стадия количественного анализа, в которой производится расчёт концентраций компонентов в анализируемой пробе.
Хроматограмма, в зависимости от поставленной цели, может служить для выполнения градуировки прибора либо для расчёта концентраций в пробе с неизвестным содержанием компонентов.
2.1 Система подготовки газов
Система подготовки газов предназначена для установки, стабилизации и измерения скорости потоков газа-носителя и дополнительных газов, питающих некоторые детекторы, а также для очистки газов. Особое значение имеют установка и стабилизация оптимального для данного анализа расхода газа-носителя, оказывающего непосредственное влияние на параметры удерживания и размеры пиков анализируемых веществ. Важно также исключить влияние колебаний расходов газа-носителя и дополнительных газов на чувствительность детекторов, чтобы не допустить связанных с этим неконтролируемых изменений параметров пиков. Кроме того, недостаточная стабильность газовых потоков часто является причиной неустойчивой нулевой линии, что затрудняет количественную обработку хроматограмм.
В систему установки и стабилизации газовых потоков входят: редуктор, РД, РРГ.
РД стабилизирует изменение давления на входе в колонку, вызванного колебаниями внешнего давления газа.
В некоторых случаях, например, при программировании температуры, необходимо поддерживать постоянные значения расхода газа-носителя через колонку, когда ее сопротивление изменяется в процессе анализа. Для этой цели используется регулятор расхода газа.
Например, при увеличении сопротивления колонки с увеличением температуры и давления РРГ работает таким образом, что начальное давление в колонке сохраняется на протяжении всего анализа неизменным.
Поскольку РРГ реагирует на изменение выходного давления, время его реакции зависит от объема газовых линий после регулятора: чем меньше этот объем, тем быстрее меняется в нем давление до порога срабатывания регулятора. Для уменьшения инерционности регулятора следует стремиться к сокращению этого объема, который складывается из объемов фильтров, манометров, дозаторов, соединительных линий и колонки. Инерционность РРГ приводит к неудовлетворительной стабилизации расхода через колонку. Отклонения текущего расхода от первоначального из-за температурных изменений сопротивления колонки может доходить до нескольких %, а восстановление расхода занимает десятки секунд.
Принципиально иным подходом к решению проблемы является использование для управления расходами газов микропроцессоров в совокупности с соответствующими измерительными и исполнительными устройствами. Созданная на этой основе автоматизированная система регулирования обладает необходимой универсальностью и достаточным быстродействием.
Очистка газовых потоков от пыли, влаги и органических соединений выполняется с помощью фильтров, заполненных достаточно активными адсорбентами (силикагель, уголь, молекулярные сита). Чистота газов особенно важна при работе с высокочувствительными ионизационными детекторами (ПИД, ЭЗД, аргоновым, гелиевым), где примеси могут являться дополнительными источниками искажения нулевой линии. Допустимый уровень загрязнения газов зависит от стабильности газовых потоков, так как колебания расхода газа могут фиксироваться детектором в виде переменного фонового потока, создаваемого примесями, и приводят к нарушению устойчивости нулевой линии.
Соединения элементов газовых линий в хроматографах обычно выполняются с помощью трубок из нержавеющей стали малого диаметра (0,5-2 мм). Уплотнение соединений осуществляется плоскими или фигурными прокладками: металлическими (медь, алюминий) или графитовыми для соединений в горячей зоне и мягкими (резина, полимерные материалы) для соединения элементов, работающих при комнатной температуре и при нагреве до 250 °C.
В простом варианте схема газовой линии в хроматографе, следующая:
Редуктор (манометр) → фильтр первичной очистки → РД → РРГ → фильтр очистки газа на входе в колонку
Герметичность газовой линии проверяют, заглушив выход газовой линии. Расход газа на РРГ должен показывать "0", либо значение от 0 до 1 (для РРГ-10), либо разница в показаниях расхода газа для ГН1 и ГН2 не должна превышать 10 мл/мин (для РРГ-11).
2.2 Выбор газа-носителя
Прежде всего, перед тем как приступить к созданию методики нам необходимо определиться с газом носителем. При выборе газа-носителя руководствуются прежде всего вязкостью последнего, а также другими свойствами, такими как кинетика массопередачи посредством диффузии, требуемая чистота и стоимость. Было доказано, что лучшим газом-носителем для газовой хроматографии является водород. Помимо водорода применяют гелий, азот, аргон.
Вязкости газов при различных температурах неодинаковы, некоторые цифры вязкости газов представлены в таблице ниже.
Таблица 2.1 – Вязкости газов при различных температурах
|
Газ |
Температура, °С | |||||||
|---|---|---|---|---|---|---|---|---|
|
0 |
20 |
50 |
100 |
150 |
200 |
300 |
400 | |
|
Н2 |
84 |
88 |
94 |
103 |
113 |
121 |
139 |
154 |
|
He |
186 |
196 |
208 |
229 |
250 |
270 |
307 |
342 |
|
Ar |
212 |
222 |
242 |
271 |
297 |
321 |
367 |
410 |
|
N2 |
166 |
176 |
188 |
208 |
229 |
246 |
279 |
311 |
|
CO2 |
138 |
147 |
162 |
185 |
205 |
229 |
268 |
– |
|
H2O пар |
128 |
147 |
166 |
201 |
235 | |||
Исходя из таблицы видно, что водород при любых температурах обладает наименьшей вязкостью, но в силу его опасных свойств не находит широкого применения.
Следует указать, что с увеличением вязкости газа-носителя перепад давления, требуемый для достижения объемной скорости потока газа-носителя через колонку, возрастает. Не существует никакого способа изменения или регулировки вязкости. Следует заметить, что водород предпочтительнее гелия, так как его вязкость ниже вязкости гелия более чем в два раза, подобным образом предпочтительнее использовать азот, а не аргон.
Все газы, используемые в газовой хроматографии в качестве подвижной фазы можно расположить в следующей последовательности:
Водород > гелий > азот > аргон или углекислый газ
Гелий выбирают в качестве газа-носителя, когда работают с детектором по теплопроводности (ДТП) для создания максимальной чувствительности и азот, когда работают с пламенно-ионизационным детектором (ПИД).
Например: для анализа компонентного состава газа, определение сероводородного состава нефти используют гелий. Для спиртового анализа используется в качестве газа-носителя азот, так же, как и в анализе жирнокислотного состава масел растительного и животного происхождения, анализе хлорорганических пестицидов и ПХБ, в анализе лекарственных и наркотических препаратов, в анализе компонентного состава бензинов. Для анализа воздуха рабочей зоны и атмосферного воздуха используется как азот, так и гелий – все зависит от конкретной методики. Для определения растворенных газов в трансформаторном масле используют в качестве газа-носителя аргон.
В некоторых случаях, в виду сложности проводимых анализов, прибегают к программированию скорости газа-носителя. Это относится, прежде всего, к капиллярным колонкам, где имеется возможность программирования скорости, потока или давления газа-носителя, так как используется регулятор РРГ-11 (о характеристиках которого мы говорили на предыдущих занятиях). При работе с насадочными колонками такую операцию осуществить пока невозможно.
Внимание! Хочется сразу предостеречь тех, кто будет пытаться улучшить качество разделения исследуемых компонентов путем оптимизации объемной скорости газа-носителя.
Следует помнить, что с увеличением линейной скорости газа-носителя при очень малых значениях этой скорости, высота эквивалентной теоретической тарелки уменьшается, проходит через минимум, а затем неограниченно увеличивается с повышением линейной скорости газа-носителя. При определенном значении скорости газа-носителя эффективность колонки максимальна. При этой объемной скорости газа-носителя степень разделения самая большая. Часто считают, что продолжительность анализа можно сильно сократить, если эксплуатировать колонку при большей скорости газа-носителя, когда эффективность колонки уменьшается незначительно. Это целесообразно делать, если эффективность Вашей колонки слишком высока для исследуемого разделения, но, если специализированная колонка предназначена для определенного анализа, лучше использовать другие методы оптимизации разделения исследуемых компонентов (например: использовать колонку меньшей длины, либо меньшего внутреннего диаметра; использовать сорбент с меньшим размером частиц; использовать программирование температуры).
Постоянное использование высокой скорости газа-носителя в процессе анализа может привести к потере фазы, после чего колонка уже не подлежит восстановлению, так как технология нанесения жидкой фазы на внутреннюю поверхность капиллярной колонки чрезвычайно сложна и требует специальных приспособлений, а потому требует замены на новую, что весьма дорого.
При проведении анализов на капиллярных и насадочных колонках часто приходится избавляться от труднолетучих компонентов, которые могут частично остаться в лайнере или в колонке, в этом случае используют не продувку колонки путем повышения скорости газа-носителя, как это можно сделать для насадочной колонки, а применяют программирование температуры, то есть конечную температуру анализа задают чуть выше температуры кипения самого высококипящего компонента. Но, не следует очень долго прожаривать колонку, длительная высокая температура также отрицательно сказывается на качестве фазы колонки.
Поскольку передача методики и обработка хроматографической информации выполняются посредством использования программы сбора и обработки хроматографической информации "Хроматэк Аналитик", то при задании режима расхода газов Вы можете встретиться с такими понятиями как поток, линейная скорость и давление газа-носителя.
Поток газа-носителя может быть охарактеризован объемной или линейной скоростью потока. Подвижная фаза протекает через колонку и независимо от того, является колонка насадочной или полой, она оказывает некоторое сопротивление потоку газа-носителя. Это сопротивление преодолевается путем подачи жидкой фазы в колонку под давлением. При этом из-за сжимаемости газа возникает профиль давлений и скоростей вдоль оси колонки.
Распределение локальной скорости газа-носителя внутри насадочной и капиллярной колонки неодинаково. В насадочной колонке, где каналы, по которым проходит газ, постоянно меняют форму и размеры, проследить распределение локальной скорости чрезвычайно сложно. Можно сказать, что скорость внутри пор частиц сорбента пренебрежимо мала, а газовый поток, окружающий частицы, хотя и ламинарный, но образует большое число стабильных завихрений в местах, доступных для газовой фазы, поэтому для насадочных колонок задается поток газа-носителя.
В полых капиллярных колонках структура потока более проста. Поперечное сечение колонки постоянно, линии потока параллельны и в центре колонки скорость потока максимальна, поэтому в капиллярных колонках проще отслеживать линейную скорость потока. Однако, при работе с капиллярными колонками можно оперировать давлением, эта величина часто используется, когда работают в режиме программирования.
К примеру, Вы работаете в условиях программирования температуры. Совершенно очевидно, что с повышением температуры, скорость газа-носителя будет уменьшаться ввиду увеличения вязкости последнего. Но, газовая схема, разработанная для нашего хроматографа, включает автоматический регулятор расхода газов и давление на входе в колонку с повышением температуры повышается, то есть проще регулировать в данном случае давление.
2.3 Дозирующие устройства
Дозирующие устройства предназначены для введения в хроматографическую колонку определенного количества анализируемой пробы.
При введении пробы должны выполняться несколько общих требований:
Состав пробы, введенной в колонку, должен быть идентичен составу анализируемой пробы, за исключением тех случаев, когда не требуется определять полный состав смеси. Нарушение идентичности состава пробы и анализируемой смеси может быть вызвано многими причинами, в частности: наличием в дозаторе непродуваемых ("мертвых") объемов, потерей части пробы при введении ее в колонку, химическими реакциями между компонентами пробы или термодеструкцией их, вызванной высокой температурой дозатора и каталитическим действием материалов дозатора, который контактирует с пробой.
При многократном введении пробы в постоянных условиях величина пробы должна быть воспроизводима (1-3%). Требования к воспроизводимости могут существенно различаться в зависимости от выбранных способов градуировки хроматографа и обработки хроматограмм, а также от требуемой точности анализа.
При введении пробы ее разбавление газом-носителем должно быть минимальным. Размывание пробы зависит от конструкции дозатора и температурного режима. Однако температура дозатора не должна быть слишком высокой, чтобы исключить возможность термической деструкции различных веществ.
Введение пробы не должно вызывать изменения установившегося режима систем хроматографа (зашкаливание нулевой линии в момент введения пробы, резкого изменения давления газа-носителя и температуры дозатора), что может быть обусловлено разгерметизацией системы при введении слишком большой пробы, изменением сопротивления линии газа-носителя при дозировании.
Величина пробы выбирается таким образом, чтобы не вызывать перегрузки колонки и с учетом чувствительности детектора, который должен четко зарегистрировать соответствие количества разделяемых веществ.
Не существует универсальных дозирующих устройств.
В зависимости от агрегатного состояния пробы, имеются различные дозирующие устройства:
Газовые краны – газообразные пробы (количественный анализ газообразных образцов часто требует точного измерения объема вводимой пробы, которая складывается из объемов дозирующей петли и газовых трактов. Современные конструкции газовых кранов комплектуются сменными дозами от 0,1 до 10 мл, при этом постоянный объем газовых трактов составляет от 0,2 до 1 мл).
Жидкие смеси в колонку вводятся специальными шприцами через термостатируемые резиновые уплотнения испарителя.
Испаритель представляет собой нагреваемый до определенной температуры металлический блок с каналом для ввода пробы и испарения жидкой пробы.
В канал подается поток предварительно нагретого газа-носителя. С одной стороны испаритель закрыт резиновой пробкой, с другой – колонкой. Иглу шприца вводят через термостатируемое уплотнение в канал испарителя, введенная проба быстро испаряется и переносится потоком газа-носителя в колонку. Для эффективного введения пробы очень важно, чтобы в канале испарителя не было непродуваемых объемов, так как это приводит к длительному ее вымыванию.
Температура испарителя влияет на начальную ширину полосы пробы в колонке и выбирается на 30-50 °C выше температуры самого высококипящего компонента смеси. Если повышение температуры испарителя приводит к увеличению эффективности разделения, значит, начальная температура испарителя была выбрана неправильно. Но, излишнее ее повышение нежелательно, так как может вызвать нежелательную деструкцию компонентов, или перегрев начальной части колонки, если та подсоединена непосредственно к испарителю.
При температуре испарителя больше 300 °C резиновое уплотнение становится менее эластичным, нарушая герметичность испарителя. К увеличению "хвостов" пиков и снижение эффективности разделения смеси приводит попадание резиновой крошки в канал испарителя.
Предпочтительнее конструкции испарителей, в которых функции канала испарения пробы выполняет сменный вкладыш, изготовленный из металла или стекла.
Стеклянные вкладыши имеют следующие преимущества:
равномерный прогрев
наименьшая способность к сорбции полярных соединений (вода, аммиак, карбоновые кислоты)
вероятность термодеструкции снижается
контроль образования налета на вкладыше и своевременная замена
Большое значение имеет внутренний диаметр вкладыша, определяющий объем канала испарения пробы.
Если объем канала испарителя намного больше, чем объем пробы, то произойдет нежелательное размывание паров анализируемой смеси и, следовательно, снижение эффективности разделения. Если объем канала испарителя намного меньше, чем объем пробы, то излишек пробы выйдет в зазор между внешней стенкой вкладыша и стенкой испарителя, что приведет к более замедленному вводу пробы.
В испаритель пробу вводят специальным шприцем на 1, 10, 50 мкл. В шприцах шток выполнен из вольфрама или нержавеющей стали.
Недостатком шприца является большой объем иглы (около 2 мкл). Часть жидкости, заключенная в игле, не выдавливается из шприца при полном введении поршня в цилиндр, но частично испаряется при введении иглы в испаритель. Количество извлекаемой из иглы жидкости зависит от глубины ввода пробы и продолжительно пребывания иглы в испарителе.
При использовании микрошприцев и наличии опыта оператора воспроизводимость введения жидких проб можно довести до 1,5-2,5%.
При переходе от одной смеси к другой шприц должен быть тщательно промыт растворителем.
Иногда требуется ввести гораздо меньшую пробу, чем это может сделать микрошприц, для этого используются делители потока:
большая часть пробы сбрасывается в атмосферу
меньшая часть попадает в колонку
Работая с делителем потока, можно решить сразу две задачи:
уменьшение величины пробы
обеспечивается эффективность ввода пробы
Для капиллярных колонок соотношение деления потока выбирается в интервале от 1:50 до 1:500. Делитель должен быть единым целым с испарителем, а может включаться дополнительно.
Для повышения эффективности перемешивания пробы и газа-носителя вкладыш испарителя заполняют стекловатой, стеклянным бисером или твердым инертным носителем, смоченным небольшим количеством 1-3% фазой типа SE-30.
Желательно, чтобы линия сброса имела температуру, равную или близкую температуре испарителя. В противном случае возможная конденсация паров высококипящих компонентов может привести к постепенному изменению соотношения расходов газа-носителя через колонку и линию сброса, вплоть до полной закупорки линии сброса.
Наиболее универсальными представляются конструкции испарителей, предусматривающих возможность работы с делителем (split) и без делителя (splitless) потока.
Такие конструкции снабжены обдувом резиновой пробки испарителя, препятствующим появлению ложных пиков.
Дозирование – одна из ответственных операций, которая осуществляется вручную, и ошибка при выполнении этой операции составляет, как правило, большую часть погрешности анализа.
В последнее время сделаны успешные попытки автоматизации дозирования жидких проб: созданы различные типы микродозаторов, позволяющих существенно повысить точность и воспроизводимость анализа.
Управление дозаторами осуществляется автоматически, по программе с компьютера.
2.4 Хроматографические колонки
Различают три основных типа аналитических колонок:
насадочные;
микронасадочные;
капиллярные.
Первые из колонок, появившиеся на этапах развития газовой хроматографии – насадочные, изготавливаются до сих пор из нержавеющей стали, стекла, фторопласта с внутренним диаметром 204 мм, длиной от 0,5-3,0 м до 6,0 м, которым придается спиральная форма.
Микронасадочные отличаются от насадочных только диаметром трубок (0,8-1,0 мм). Длина колонок не превышает 2 м. Чаще хроматографы комплектуются насадочными колонками из нержавеющей стали, но если анализируемые вещества не стойкие к каталитическому окислению или корродированию, то используют стеклянные колонки. Колонки из фторопласта хорошо зарекомендовали себя при анализе на содержание малых примесей высокополярных соединений (вода, аммиак и другие), однако их нельзя эксплуатировать при температуре выше 90-100 °C.
Капиллярные колонки выполняются из нержавеющей стали, стекла, кварца; внутренний диаметр 0,25-0,50 мм; длина 10 (20) – 100 (200) м. Неподвижная фаза в виде тонкой пленки нанесена на внутренние стенки колонки, то есть колонка остается полой. Поток газа движется в колонке с большой линейной скоростью, не встречая значительного сопротивления. Поэтому для обеспечения необходимых расходов газа-носителя через капиллярную колонку оказывается достаточным примерно такое же входное давление, что и при работе с насадочными колонками.
Замечательной особенностью капиллярных колонок является весьма высокая эффективность (до нескольких тысяч тарелок на 1м). Эффективность работы капиллярной колонки в значительной мере определяется чистотой и однородностью внутренней поверхности капилляра.
В 70-е г.г большое распространение получили стеклянные капиллярные колонки (анализ термически и каталитически неустойчивых, а также высокополярных соединений). Стекло – наиболее доступный и дешевый материал. Однако низкая механическая прочность стеклянных колонок значительно затрудняет работу с ними.
В конце 70-х г.г. сформировалось новое направление – использование тонкостенных кварцевых капиллярных колонок, внутренний диаметр которых составляет 0,05-0,32 мм; толщина фазы – 40-70 мкм; толщина защитного слоя – 15-30 мкм. Низкая остаточная адсорбционная активность сочетается в них с высокой механической прочностью на изгиб. В качестве материала для изготовления колонок используют очищенный кварц или синтетически плавленый диоксид кремния.
Долговечность и гибкость колонок достигается путем нанесения лака (полиимид) сразу после вытяжки, защищающей поверхность от внешних воздействий, и допускает длительную работу при 250-300 °C. Кварцевая капиллярная колонка допускает изгиб на диаметр менее 10 мм.
Важным ограничением в использовании классических капиллярных колонок малого диаметра (0,25-0,32 мм), толщиной пленки около 1 мкм для определения примесей – их невысокая емкость, теперь преодолены использованием широких (внутренний диаметр 0,53 мм) капиллярных колонок, с толщиной слоя до 5-8 мкм. Рабочая емкость капиллярных колонок приближена к насадочным, в следствие чего можно работать без деления потока.
2.5 Выбор колонки для анализа
Итак, вы определились с новой аналитической задачей и желаете решить ее с помощью газовой хроматографии и первый вопрос, который у вас возникает: "Какую выбрать фазу?"
От правильного выбора фазы колонки во многом определяется качество разделения анализируемых компонентов. Этот этап наиболее сложный в постановке анализа, так как требует от аналитика глубоких знаний о природе анализируемых компонентов, неподвижных и жидких фаз.
Поскольку анализы могут проводиться как на насадочной, так и на капиллярной колонках, поэтому выбор носителей и фаз для хроматографирования несколько отличается друг от друга.
Для насадочных колонок спектр возможных фаз гораздо больше, чем для капиллярной колонки, также, как и носителей, кроме того, процедура нанесения жидкой фазы на твердый носитель для насадочных колонок значительно проще, то возможно использование комбинированных фаз, нанесенных на твердый носитель.
Если говорить о капиллярных колонках, следует прежде всего отметить, что они делятся на:
WCOT – колонки – полые капиллярные колонки, в которых тонкая пленка неподвижной фазы нанесена непосредственно на внутреннюю поверхность. Обладает высокой эффективностью по отношению к трудноразделяемым смесям, состоящим из большого числа компонентов. Промышленность выпускает в основном колонки с внутренним диаметром от 0,05 до 0,53 мм. Слой неподвижной фазы толщиной от 0,1 до 0,8 мкм.
PLOT – колонки – кварцевые капиллярные колонки, на внутренние стенки которых нанесен слой адсорбента Al2O3/KCl, молекулярные сита или пористые полимеры (близкие по составу к Porapak Q).
SCOT – колонки – капиллярные колонки, на внутренних стенках которых нанесен слой носителя с неподвижной фазой.
В настоящее время промышленностью выпускаются 9 основных типов жидких фаз, наиболее часто используемых в WCOT – колонках:
Неполярные – OV-1; SE-30; OV-101, SP-2100 (100% диметилполисилоксан); SE-52; OV-23; SE-54
Cредней полярности – OV-1701; OV-17; OV-225; OV-210
Полярные – OV-101, SP-2100 (ПЭГ, модифицированный терефталевой кислотой); Карбовакс 20М
При выборе колонки поступают следующим образом:
Испытывают имеющуюся колонку
Обращаются за советом к коллегам
Проводят литературный поиск
При выборе неподвижной фазы важно помнить, что она должна обладать избирательностью по отношению к исследуемым компонентам, термостабильностью и обеспечивать высокую эффективность колонок.
Избирательность оценивается возможностью разделения двух выбранных веществ и количественно оценивается относительным удерживанием этих соединений.
Термостабильность характеризуется:
Уносом неподвижной фазы из колонки за счёт истирания, разложения, испарения;
Помехи на хроматограмме, вызванные фоном паров
К выбираемой жидкой фазе также предъявляется ряд требований:
Малая летучесть при рабочих температурах;
Селективность;
Химически инертная по отношению к исследуемому образцу. Летучесть и термическая стабильность уменьшаются с повышением молекулярной массы вещества. Верхний температурный предел зависит не только от природы жидкой фазы и рабочих температур, но и от природы твердого носителя, от количества нанесенной жидкой фазы, от типа колонок;
Низкая вязкость при рабочей температуре;
Хорошая растворимость в наиболее доступных распространенных растворителях
Чтобы точнее определиться в выборе фазы для колонки в специальной литературе по хроматографии даются таблицы полярностей наиболее широко применяемых фаз и те группы веществ, которые могут быть определены с использованием данной фазы. Полярность неподвижных жидких фаз определяется по классификации Мак-Рейнолдса.
И еще одно очень важное условие выбора жидкой фазы: подобное растворяется в подобном. Не следует проводить разделение полярных компонентов на неполярных жидких фазах и наоборот.
2.5.1 Выбор твердого носителя и неподвижной жидкой фазы для колонки
Для того, чтобы ваша колонка работала наиболее эффективно, необходимо правильно подобрать твердый носитель (ТН) и неподвижную жидкую фазу (НЖФ), которые наилучшим образом подойдут для хроматографирования исследуемой пробы. Мы рассмотрим лишь некоторые, наиболее часто используемые ТН и НЖФ.
В совокупности ТН и НЖФ представляют собой фазовую систему. Обычно для работы выбирают носитель неподвижной жидкой фазы с размером частиц 0,1-0,3 мм, но в некоторых случаях для лучшего разделения использую носители с меньшим или большим диаметром частиц, но, как правило, выполняется условие: внутренний диаметр колонки должен быть равен 8-10 диаметрам носителя неподвижной жидкой фазы.
Используемые неподвижные фазы могут быть подразделены на три основные группы:
Адсорбенты с очень большой удельной поверхностью (50-1000 м2/г): силикагель, оксид алюминия, молекулярные сита, активный уголь и графитированная сажа.
Нейтральные или инертные носители обычно получают из диатомитовых материалов, иногда из полимеров. Их получают путем нанесения на поверхность материалов жидкости с очень низким давлением пара и высокой термической стабильностью в условиях использования. Имея под рукой широкий спектр неподвижных носителей и жидких фаз можно регулировать селективность разделения компонентов смеси.
Для проведения сложных разделений очень успешно используются адсорбенты с нанесенным на них малым количеством жидкости с низким давлением пара. Этот метод называется газоадсорбционной хроматографией.
Подвижной фазой служит инертный газ (гелий, азот, аргон).
Среди твердых носителей выделяют следующие группы:
Диатомитовые носители
Молекулярные сита
Полимерные адсорбенты
Углеродные адсорбенты
Диатомитовые носители делятся на два типа: I тип носителей помимо высокой механической прочности и меньшей хрупкости, характеризуется также значительно более высокой по сравнению с носителями II типа эффективностью разделения и лучшей способностью к пропитке жидкой фазой. Однако имеются и ряд недостатков: вследствие большей поверхности (4м2/г по сравнению с 1 м2/г для носителей второго типа) носители первого типа значительно более реакционноспособны и являются лучшими адсорбентами.
К носителям первого типа относятся: Хромосорб A,G,W; Порохром 1,2,3; Хроматон N, N-Супер; Инертон.
К носителям второго типа относятся: Хромосорб красный; Хромосорб P,R; Сферохром 2,3.
Таблица 2.2 – Свойства некоторых диатомитовых носителей
|
Название носителя |
Химический состав |
Удельная поверхность, |
Насыпная плотность, |
Макс. степень пропитки, % |
Тип |
|---|---|---|---|---|---|
|
Хромосорб W |
Кремниевая кислота с переменным количеством оксидов Al, Fe, Mg, Ca, H2O |
1…3 |
0,24 |
30 |
II |
|
Хромосорб P |
– |
4 |
0,48 |
30 |
I |
|
Хромосорб R |
– |
4…4,8 |
– |
– |
– |
|
Хромосорб G AW,DMCS |
– |
0.5 |
0.58 |
5 |
II |
|
Хроматон N |
Кремниевая кислота с переменным содержанием оксидов Fe, Al, Ca, MgO, H2O |
1 |
0.24 |
I | |
|
Инертон |
– |
0,4…0,6 |
II | ||
|
Сферохром 2,3 |
– |
4…15 |
II | ||
|
Порохром– 1 |
– |
0,1…1,5 |
II | ||
|
Порохром-2 |
– |
0,5…2 |
II | ||
|
Полихром-1 |
Полимер |
3…6 |
0,7 |
||
|
Хромосорб Т |
4,8 |
||||
|
Оксид алюминия |
Al2O3+примеси |
Активность и другие свойства:
Хромосорб W. Возможны реакции с О-содержащими соединениями; "хвосты" с насыщенными спиртами; высокое газовое сопротивление.
Хромосорб P. Очень активен.
Хромосорб R. То же, малое газовое сопротивление.
Хромосорб GAW,DMCS. Еще менее активен, устойчив к скалыванию, однороден, воспроизводим.
Хроматон N. Малоактивен.
Полихром-1. Не проявляет ни каталитической, ни адсорбционной активности.
Оксид алюминия. Высокая каталитическая и адсорбционная активность.
2.5.2 Углеродные адсорбенты
Графитированная термическая сажа представляет собой непористый, инертный и устойчивый к высокой температуре адсорбент с высокой удельной поверхностной энергией. Легко модифицируется различными жидкими и твердыми фазами, а это позволяет проводить селективное разделение самых различных соединений. Малопригодна для газового анализа при обычной температуре из-за малой удельной поверхности (6-12 м2/г). Для такого анализа применяется сажа с удельной поверхностью около 100 м2/г, выпускаемая под названием Карбопак В.
Таблица 2.3 – Основные характеристики углеродных адсорбентов
|
Адсорбент |
Карбопак А |
Карбопак В |
Карбопак С |
Сферон-6 (графон) |
|---|---|---|---|---|
|
Изготовитель |
Супелко |
– |
– |
Phase separation |
|
Sa, м2/г |
15 |
100 |
9 |
87 |
|
Насыпная плотность, г/см3 |
0,66 |
0,34 |
0,66 |
|
|
Диаметр первичных частиц, мм |
150…225 |
31 |
200 |
6 |
|
Рекомендуемая область применения |
Газовый анализ, НМС, накопление микроколичеств |
Разделение структурных и стереоизомеров | ||
Молекулярные сита природные и искусственные цеолиты представляют собой кристаллические алюмосиликаты с трехмерной структурой из тетраэдров SiO4 и AlO4 M2/nO*Al2O3*xSiO2*yH2O (M-щелочной или щелочноземельный металл, n– его степень окисления), объединенных в те или иные полиэдры. Имеют большую внутреннюю поверхность 700-800 м2/г. Адсорбционные свойства цеолитов определяются стерическими, энергетическими, кинетическими эффектами. В газовой хроматографии применяются в основном синтетические цеолиты 5А, 10Х, 13Х. Поскольку молекулярные сита этой группы отличаются большой удельной поверхностью, их используют преимущественно для разделения низкокипящих газов. Кроме того, молекулярные сита можно использовать для выявления в смесях высококипящих углеводородов.
Полимерные адсорбенты – сферические высокопористые частицы, нерастворимые в кислотах, основаниях и органических растворителях. Особенно важным свойством этих полимеров является их гидрофобность, обусловленная отсутствием гидроксильных групп. Малое сродство к соединениям, содержащим ОН-группы, имеет следствием относительно малое время удерживания и симметричные пики воды, спиртов, гликолей, карбоновых кислот, аминов и др. Такие важные свойства как высокая химическая и геометрическая гомогенность, механическая прочность, большая емкость, гидрофобность, способствовали тому, что получили широкое распространение.
Основной областью применения этих материалов является газовый анализ, включая анализ природных газов в присутствии водяного пара.
Таблица 2.4 – Основные характеристики адсорбентов типа Порапак
|
Тип aдсорбента |
Матрица |
Sa, м2/г |
D50, нм |
Полярность |
Tmax, °С |
|---|---|---|---|---|---|
|
Порапак N |
Сти-ДВБ с винилпирролидоном |
250…350 |
Среднеполярная |
190 | |
|
Порапак R |
Сти-ДВБ с винилпирролидоном |
450…600 |
7…10 |
Слабополярная |
250 |
|
Порапак Q |
Этилвинилбензол-ДВБ |
500…600 |
7,5…10 |
Неполярная |
250 |
Таблица 2.5 – Область применения адсорбентов типа Порапак
|
Тип aдсорбента |
Область применения |
|---|---|
|
Порапак N |
Определение формальдегида в водных растворах, разделение NH3, CO2, H2O; определение C2H2 в углеводородах С2. |
|
Порапак R |
Разделение простых и сложных эфиров, нитрилов, нитросоединений, определение воды в неорганических соединениях, HCl, Cl2. |
|
Порапак Q |
Разделение алканов, спиртов, соединений серы, оксидов азота, определение органических соединений в воде. |
Селективность колонки определяется природой НЖФ. Система для характеристики НЖФ была предложена Мак-Рейнольдсом. Им было охарактеризовано порядка 200 НЖФ и предложена шкала "полярности" НЖФ. Оказалось, что селективности многих НЖФ, используемых в газовой хроматографии с насадочными колонками, очень близки, поэтому был составлен перечень наиболее предпочтительных фаз, что облегчает выбор колонки для исследования.
Таблица 2.6 – Рекомендации по выбору неподвижных жидких фаз
|
Торговое название |
Структура |
Применение |
Предел температуры, °С |
|---|---|---|---|
|
Apiezon L |
Метилфениловые эфиры |
1,2,3,4 |
300 |
|
SE-30 |
Метилсиликон |
1,2 |
300 |
|
OV-101 |
Метилсиликон |
1,2,3 |
250 |
|
SP-2100 |
Метилсиликон |
1,2,3 |
350 |
|
OV-17 |
Фенилсиликон |
1,2,3 |
375 |
|
OV-210 |
Фторалкилсиликон |
1,2,3 |
275 |
|
XE-60 |
Нитрилсиликон |
1,2,3 |
250 |
|
OV-225 |
Нитрилсиликон |
1,2,3 |
275 |
|
Carbowax 20M |
Полиэтиленгликоль |
1,2,3,4 |
225 |
|
Carbowax 6000 |
Полиэтиленгликоль |
2,3,4 |
200 |
|
Carbowax 1500 |
Полиэтиленгликоль |
2,3,4 |
175 |
|
Reoplex 400 |
Полиэтиленгликольадипат |
2,3,4 |
220 |
алканы
алкены, алкины, ароматические углеводороды, эфиры, альдегиды, кетоны, третичные амины, нитрилы, азаарены, тиаарены, тиолы
металлорганические соединения
вода, карбоновые кислоты, спирты, первичные и вторичные амины, сложные эфиры
В таблице приведены максимальные температуры для чистых НЖФ, но при нанесении НЖФ на твердый носитель, ее свойства меняются, поэтому при кондиционировании колонок максимальную температуру необходимо выставлять на 15-20 °C ниже указанной в таблице.
В случае отсутствия подходящего сорбента можно использовать:
специально приготовленные фазы
например, 10% НТПН на Хроматон N-AW-HMDC 0,16-0,20 мм для анализа толуола, бензола, м‑,п-ксилол, о-ксилол, стирол в промышленном воздухе;
10% ДНФ на Хроматон N-AW-HMDC 0,16-0,20 мм для анализа спиртов в биологических жидкостях и др.
колонки со смешанными фазами
последовательно соединенные колонки с различными НЖФ, например – колонки с ТЗК, модифицированным вазелиновым маслом и дибутилфталатом для анализа компонентного состава сжиженного газа.
Ввод пробы в капиллярный и насадочный испаритель осуществляется как шприцем, так и автоматическими дозирующими устройствами. Конечно, ввод пробы с помощью автоматического устройства намного удобнее, так как реализуется быстрый ввод пробы, который нельзя осуществить, работая шприцем, благодаря чему увеличивается воспроизводимость анализа.
2.6 Выбор температурного режима
Во многих случаях аналитик должен определить состав сложных смесей, содержащих много компонентов с очень различающимися давлениями паров, энергиями испарения и полярностями. Невозможно найти оптимальную температуру, при которой бы в изотермическом режиме одинаково хорошо делились и легкие, и тяжелые компоненты.
Если использовать низкие температуры, то будут хорошо делиться низкокипящие компоненты, а тяжелые компоненты будут давать широкие плоские пики, трудные для детектирования и количественной обработки.
Либо, если мы задаем высокую температуру анализа, тяжелые компоненты будут делиться хорошо, но тогда мы не разделяли бы легкие компоненты.
Так как в этом случае нет приемлемой температуры, то необходимо искать другой подход – либо применение нескольких колонок с переключением колонок во время анализа, либо газовая хроматография с программированием температуры.
Есть одно условие при выборе температуры колонки:
Температура колонки должна быть на 50 °С выше, чем температура кипения основного компонента смеси, а если в пробе несколько компонентов, то на 50 °С выше самой высокой температуры кипения этих веществ.
На данном этапе в вашу задачу входит:
получение общего представления о сложности смеси
сформировать мнение о способности общепринятых фаз обеспечить достижение требуемого разделения
Если в результате этих экспериментов Вам не удалось разделить интересующие вещества, то бесполезно экспериментировать с длиной колонки, какой бы длины колонку Вы не взяли, разделения не получите, в этом случае необходимо менять фазу колонки. Или можно попробовать капиллярную колонку, если до сих пор анализ проводился на насадочной колонке.
В том случае, когда анализируются смеси компонентов одной природы достаточно работать в изотерме.
Например:
анализ ароматических соединений в воздухе рабочей зоны (бензол, толуол, м-п-ксилол, о-ксилол, стирол);
анализ спиртов в биологических жидкостях;
анализ галогенсодержащих в воде (парофазный анализ);
анализ пестицидов;
анализ ПХБ;
анализ наркотических веществ.
Если нам необходимо провести анализ сложных смесей, то тогда используют программирование температуры. Программирование температуры обеспечивает возможность элюирования всех компонентов за приемлемое время при одновременном достижении хорошей степени разделения и довольно малых пределов детектирования.
Например:
жиры или сложные эфиры жирных кислот;
производные аминокислот;
анализ бензинов;
анализ спиртов;
анализ наркотиков;
определение хлорорганических пестицидов;
анализ ароматических соединений в воде;
анализ спиртов в биологических жидкостях;
анализ природного, попутного и сжиженного газов;
анализ серосодержащих компонентов в нефти.
Единственной проблемой в данном случае является выбор оптимальной начальной температуры и скорости программирования. Конечная температура чаще всего устанавливается равной температурному пределу используемой неподвижной фазы. Однако, в зависимости от характера анализируемых компонентов конечную температуру необязательно задирать слишком высоко, дабы исключить испарение фазы.
Хочется отметить, что с развитием хроматографической техники, воспроизводимость анализа в условиях программирования температуры стала достаточно высокой благодаря наличию: автоматического регулятора расхода газа, специального материала для термостата колонок, линейных программаторов температур.
Как можно отметить, природа неподвижной жидкой фазы колонки играет большую роль при выборе температурного режима, так как именно от ее природы зависит задание максимально возможной температуры анализа. Давление пара неподвижной фазы должно быть низким, чтобы исключить испарение фазы, загрязнение детектора и возможную перегрузку детектора.
Чтобы избежать возможных последствий повторяемых тепловых ударов, испытываемых колонкой, наполнитель перед использованием следует тщательно прокондиционировать. Это можно сделать выдерживанием колонки в изотермическом режиме при температуре намного выше температурного предела, наблюдаемого при программировании, в постоянном потоке газа-носителя до тех пор, пока не стабилизируется нулевая линия. Иногда достигают удовлетворительного результата применением последовательных циклов программирования температур. Более того, поступенчатое кондиционирование наиболее благоприятно для неподвижной жидкой фазы колонки.
2.7 Детекторы
Хроматографический детектор представляет собой устройство, предназначенное для обнаружения и количественного определения, выходящих из колонки в потоке газа-носителя компонентов анализируемой смеси. Регистрация компонентов осуществляется за счёт преобразования в электрический сигнал изменений физических или физико-химических свойств газового потока, выходящего из хроматографической колонки.
Комплект современного газового хроматографа содержит 4-6 детекторов.
Наибольшее распространение получили в силу универсальности, превосходных характеристик и высоких эксплуатационных качеств, детекторы ПИД и ДТП. Кроме того, широко используются селективные детекторы: ЭЗД, ПФД.
Детекторы подразделяются на интегральные и дифференциальные.
Интегральный детектор – регистрирует изменение во времени суммарного количества выходящего из колонки компонента, например, их общий объем или количество титрующегося раствора, израсходованного на нейтрализацию анализируемого вещества.
Хроматограмма получается в виде ступеней, каждая из которых по высоте пропорциональна количеству компонента, прошедшего через детектор за время t2-t1 (см. рис.3.4.1). Эти детекторы имеют весьма ограниченное применение из-за большой инерционности и недостаточной универсальности.
b – измеряет мгновенную концентрацию или массовую скорость вещества в потоке газа-носителя. Хроматограмма представляет собой ряд пиков, причем количество каждого компонента пропорционально площади А соответствующего пика.
При детектировании возможны два варианта взаимодействия молекул анализируемого вещества с чувствительным элементом детектора:
процесс, разрушающий молекулы при регистрации (повторная регистрация невозможна).
процесс, в результате которого не утрачивается возможность повторной (многократной) регистрации тех же молекул.
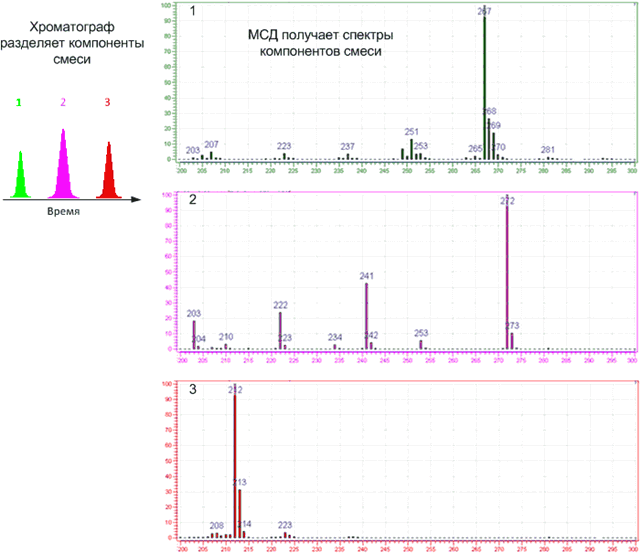
Рисунок 2.3 – Интегральная хроматограмма
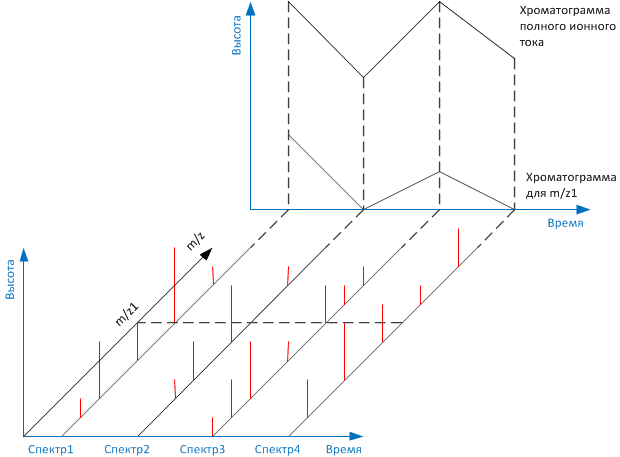
Рисунок 2.4 – Дифференциальная хроматограмма
В свою очередь, детекторы делят на потоковые и концентрационные.
При использовании потокового детектора все количество анализируемого компонента успевает однократно зарегистрироваться вне зависимости от скорости пропускания, тогда как в концентрационном детекторе от скорости зависит число актов регистрации каждой молекулы и чем больше скорость, тем меньше актов взаимодействия при одном и том же числе молекул.
Типичным примером потокового детектора является ПИД (сгорание органических соединений).
ДТП – концентрационный детектор.
При измерении площадей пиков потоковые детекторы более предпочтительны из-за независимости их показаний от колебаний давления и расхода.
2.7.1 Характеристики детекторов
Основными характеристиками детекторов является чувствительность и предел детектирования, линейность, быстродействие, селективность.
Чувствительность – связывает сигнал с измеряемой концентрацией и определяет аналитические возможности хроматографа в целом. От чувствительности зависит выбор величины пробы и возможность использования различных типов колонок.
Применение высокочувствительных детекторов весьма желательно, так как позволяет уменьшить величину вводимой пробы, что улучшает качество разделения компонентов анализируемой смеси.
Весьма важной величиной, характеризующей предел чувствительности, является минимальная концентрация анализируемого вещества в потоке газа-носителя, которая может быть зарегистрирована. Для этого нужно знать, какое наименьшее значение сигнала детектора можно измерить, учитывая уровень флуктуационных шумов нулевой линии прибора.
Минимальным сигналом Еmin, поддающимся измерению, принято считать сигнал, амплитуда которого вдвое превышает уровень шумов δ:
\[E_{\min} = \ 2 \times \delta\]
Рисунок 2.5 – Наименьший детектируемый полезный сигнала
Концентрация анализируемого вещества, вызывающего этот сигнал, для концентрационного детектора:
\[C_{\min} = \frac{Е_{\min}}{R_{с}}\ = \ \frac{2\delta}{R_{с}}\]
\[R_{с} = \frac{АVF}{qw}\]
где:
А – площадь пика
V – чувствительность
F – скорость газа-носителя, мл/с
q – масса компонента
w – скорость ленты, см/с
Для потокового детектора:
\[C_{\min} = \ \frac{2\delta}{RjF}\]
\[Rj\ = \frac{AV}{qw}\]
Величина Сmin называется пределом детектирования и является весьма важной характеристикой, поскольку она позволяет оценить предельные возможности детектора. Предел детектирования наиболее часто выражают в мг/мл; мл/мл; %об.; ppm.
В повседневной практике часто путают понятия "чувствительность" и "предел детектирования", понимая под чувствительностью минимальные концентрации, определяемые детектором.
Графически это можно выразить:
Рисунок 2.6 – Одинаковый уровень шума
Рисунок 2.7 – Различный уровень шума
Чувствительность характеризуется наклоном зависимости "сигнал детектора – концентрация вещества", а предел детектирования – отрезком на оси абцисс, соответствующим точке пересечения градуировки с ординатой, равной минимальному сигналу, доступному для измерения (двойной уровень шума 2δ).
Из этого следует, что из двух детекторов с одинаковым уровнем шумов меньшим пределом детектирования будет обладать детектор с большей чувствительностью. Однако это не значит, что детекторы с большей чувствительностью всегда способны определять меньшие концентрации, то есть имеют меньший предел детектирования. Вполне реальны случаи, когда благодаря низкому уровню шумов меньший предел детектирования будет соответствовать детектору с меньшей чувствительностью.
Поэтому сопоставление возможностей детекторов в отношении регистрации малых концентраций веществ следует производить, сравнивая предел детектирования.
Предельные возможности хроматографа в отношении измерения малых концентраций могут быть расширены двумя независимыми путями: увеличением чувствительности и уменьшением шумов.
Следует подчеркнуть, что предел детектирования соответствует концентрации вещества в газе-носителе, создаваемом в детекторе, а не концентрации анализируемых веществ в пробе при введении в колонку.
Учитывая процесс размывания пробы, нужно иметь ввиду, что практически измеренная минимальная концентрация вещества в пробе, по крайней мере, в 5-10 раз выше предела детектирования.
Весьма важной характеристикой детектора является линейность – пропорциональность между концентрацией анализируемого вещества в потоке газа-носителя на выходе из колонки и сигналом детектора.
Если измерить зависимость "сигнал детектора – концентрация вещества", то есть снять градуировочную зависимость, то прямолинейный участок этой зависимости определяет линейную область детектирования.
Рисунок 2.8 – Диапазон линейности детектора
Отклонение от прямолинейной зависимости свидетельствует об отклонении от линейности. Диапазон линейности представляет собой интервал концентраций от предела детектирования до концентраций, при которых наблюдается значительное отклонение (3-5%) от пропорциональности и определяет отношение максимума концентрации к пределу детектирования. В пределах диапазона линейности чувствительность детектора не зависит от концентрации.
Количественный анализ в условиях нелинейности детектора требует более детальной градуировки в диапазоне рабочих концентраций анализируемых веществ. Такая градуировка связана со значительными экспериментальными трудностями – точной дозировкой пробы и тщательным воспроизведением условий хроматографического анализа.
Существенное влияние на четкость регистрации результатов разделения оказывает также эффективный объем чувствительности. Он должен быть возможно меньшим по сравнению с объемом газа-носителя, в котором распределяется анализируемый компонент, выходящий из аналитической колонки, в противном случае может происходить дополнительное размывание или смещение пиков компонентов. Это особенно важно для микронасадочных и капиллярных колонок, где используют зачастую ПИД.
В зависимости от цели анализа к детектору могут предъявляться противоположные требования. Например, при установлении состава сложной смеси требуется, чтобы детектор регистрировал как можно больше пиков, причем желательно с одинаковой чувствительностью. И наоборот, большое число пиков иногда мешает определять необходимые для анализа компоненты, тогда используют селективные детекторы.
Применение селективных детекторов позволяет проводить анализ микропримесей, причем бывает единственно возможным. Также позволяет предварительно классифицировать компоненты исследуемой смеси и получить данные о природе анализируемых соединений.
Селективность детектора определяют двумя способами:
по отношению чувствительности детектора к одному и тому же веществу:
\[S\ = \ \frac{R1}{R2}\]
по отношению чувствительности одного детектора к двум веществам
\[S\ = \ \frac{Rа}{Rв}\]
При этом детектор считается селективным, если его чувствительность для двух веществ не меньше, чем на порядок.
Для качественного анализа, в частности для групповой классификации или классификации веществ, то есть возможности избирательного детектирования, предпочтителен второй способ.
Для характеристики свойств селективного детектора чаще применяются данные первого способа расчёта.
2.7.2 Детектор по теплопроводности (ДТП)
Принцип действия: изменение температуры нагретых нитей в зависимости от теплопроводности окружающего газа, которая зависит от его состава.
ДТП (катарометр) измеряет различие в теплопроводности чистого газа-носителя и смеси газа-носителя с веществом, выходящим из хроматографической колонки. Поэтому наибольшая чувствительность может быть получена в том случае, когда теплопроводность анализируемого вещества сильнее отличается от теплопроводности газа-носителя. Большинство органических веществ имеют низкую теплопроводность, и для их анализа лучше использовать газ-носитель с возможно более высокой теплопроводностью. Такими газами является водород и гелий, но так как гелий дорогой, а водород опасен, часто используют азот, аргон, углекислый газ или воздух, но ДТП с ними работает хуже.
ДТП представляет собой металлический блок, в цилиндрические отверстия которого вставляются чувствительные элементы – металлические спирали из проволоки. Камеры детектора через входной и выходной канал продуваются газом-носителем.
Чувствительные элементы изготавливаются в виде спиральных нитей диаметром 0,025-0,125 мм из материала с высоким температурным коэффициентом сопротивления (вольфрам, платина). Нити нагреваются постоянным током до температуры выше температуры блока. Например, при использовании гелия и токе в 200 мА температура нитей с сопротивлением около 50 Ом примерно на 1000С выше температуры блока детектора.
Для получения дифференциального сигнала через одну камеру детектора (измерительную) проходит газ, выходящий из хроматографической колонки, через другую (сравнительную) – чистый газ-носитель. Нагретые чувствительные элементы в камерах обдуваются потоком газа-носителя, и их сопротивление приобретает определенное значение. При прохождении через детектор бинарной смеси, в измерительной ячейке нарушается теплообмен. При нарушении условий теплообмена изменяется температурная чувствительность элемента и, как следствие, его сопротивление. Различие сопротивлений чувствительных элементов является функцией мгновенной концентрации компонентов в газовом потоке и измеряется с помощью моста Уинстона.
При выборе рабочих условий для достижения большей чувствительности необходимо повысить ток моста, снизить температуру детектора и газ-носитель выбрать с наивысшей теплопроводностью.
Предельно допустимым считается ток, который при выбранных рабочих условиях детектора нагревает чувствительные элементы до 600-7000С. Поэтому с увеличением молекулярной массы газа-носителя (уменьшением теплопроводности) предельно допустимый ток становится меньше.
2.7.3 Принципы ионизационных детекторов
Ионизационные методы обеспечивают наибольшую чувствительность и широко применяются для определения малых количеств анализируемых веществ.
Основа метода: зависимость электрической проводимости ионизированной газовой среды от ее состава.
Сигналом ионизационных детекторов является изменение ионизационного тока (электрический ток, создаваемый между электродами детектора всеми заряженными частицами в газе), вызванного введением в детектор анализируемого вещества.
Ионный ток в детекторе возникает под воздействием какого-либо источника ионизации (радиоактивного изотопа, пламени, разряда, фотоионизации, электронной и ионной эмиссии) и электрического поля (разности потенциалов) между электродами детектора.
В любой момент времени в детекторе достигается равновесие, характеризующееся тем, что скорость образования заряженных частиц (ионов, электронов) равна сумме скоростей рекомбинации и сбора заряженных частиц на электродах детектора. Скорость сбора определяет ток детектора. В ионизационных детекторах создаются такие условия, при которых либо плотность (концентрация) заряженных частиц, либо скорость переноса их в электрическом поле зависит от состава газа.
Рисунок 2.9 – Вольт-амперная характеристика ионизационных детекторов
I – участок неполного сбора заряженных частиц.
II – участок насыщения.
III – участок вторичной ионизации.
I – слабое поле (неполный сбор заряженных частиц, значительная часть успевает рекомбинироваться). Скорость зарядов определяется подвижностью, которая численно равна скорости, приобретаемой зарядом в поле напряжением 1 В/см. Подвижность пропорциональна величине заряда и обратно пропорциональна массе частиц.
Если введение анализируемого вещества вызывает увеличение рекомбинаций или существенное снижение подвижности, ток детектора падает, и это снижение тока регистрируется на хроматограмме как пик данного вещества.
На этом принципе работает ЭЗД.
II – участок насыщения (отсутствие рекомбинаций и полный сбор всех образующихся зарядов). Ионный ток определяется только скоростью образования зарядов. Сигналом детектора, работающего на этом участке, является увеличение тока, вызванное значительным увеличением скорости образования заряженных частиц вследствие ионизации анализируемого компонента, поступающего в детектор. При этом ионизация чистого газа-носителя должна отсутствовать и уровень фонового тока может быть весьма малым.
Типичным примером такого детектора является ПИД, в котором водородное пламя служит источником ионизации органических соединений; ток насыщения увеличивается пропорционально количеству вещества, поступающего в детектор.
III – участок высокой напряженности электрического поля.
Насыщение увеличивается за счёт размножения зарядов (вторичной ионизации) при введении в детектор анализируемого вещества. При ионизации газа-носителя обеспечивается постоянная скорость образования зарядов: А→А+ + e. Освобожденные электронов малых энергий разгоняются сильным полем и при соударениях с атомами газа-носителя сообщают им энергию, переводящую их в возбужденное состояние: А + e → А*. Полный сбор электронов и ионов, возникающих в результате первичной ионизации газа-носителя, создает фоновый ток детектора.
2.7.4 Пламенно-ионизационный детектор (ПИД)
Преимущества данного детектора: высокая чувствительность к органическим соединениям, широкий линейный диапазон, сравнительно малая зависимость рабочих параметров от конструкции и внешних условий, безинерционность и отсутствие жестких требований к стабильности электрического питания.
Детектор представляет собой камеру, в которой поддерживается водородное пламя. В камеру вводят водород и кислород: водород в смеси с газом-носителем через канал горелки, а воздух – через другой канал, и вся смесь равномерно распределяется. Горелка является одним из электродов и изолирована от корпуса детектора и соединена с источником стабилизированного напряжения. Второй электрод расположен над горелкой.
Поскольку в пламени чистого водорода число ионов мало, сопротивление межэлектродного газового пространства очень велико (1014 – 1013 Ом) и ток детектора весьма мал (10-12 – 10-11А). Этот ток, возникающий за счёт ионизированных примесей, содержащихся в газе-носителе, воде, кислороде, является постоянным фоновым током детектора. При внесении с газом-носителем анализируемых органических веществ число ионов в пламени резко увеличивается, сопротивление падает и во внешней цепи детектора регистрируется соответствующее возрастание ионного тока.
Механизм ионизации: в нижней части пламени (у среза горелки) происходит термическая деструкция органических молекул. Окисление продуктов деструкции сопровождается хемиионизацией, при которой вся энергия химической реакции окисления не распределяется в окружающей среде, а направлена на ионизацию:
\[CH\ + \ O\ \rightarrow \ CHO^{+} + \ e\]
\[CHO + \ H_{2}O\ \rightarrow \ H_{3}O^{+} + \ e\]
Ионы гидроксония обуславливают электрическую проводимость пламени. Механизм ионообразования объясняет пониженную чувствительность к органическим соединениям, уже имеющим окисленные атомы С. Чувствительность к соединениям, содержащим только окисленные атомы С, мала или отсутствует (например, муравьиная кислота).
Относительную молярную чувствительность ко многим органическим соединениям можно охарактеризовать эффективным углеродным числом (ЭУЧ).
Вклад каждого углеродного атома в ЭУЧ в молекуле алкана равен 1. Чувствительность ПИД обычно к разветвленным углеводородам выше, чем к их линейным изомерам.
Предел детектирования ПИД при малых уровнях флуктуационного фонового тока и при использовании современных электрометров, способны измерять токи до 10-12, а на полную шкалу, может достигать 10-9 мг/с. Это соответствует минимальному содержанию вещества 5×10-8 (об. %) при скорости газа-носителя 30 мл/мин и молекулярной массе вещества 100.
Современные детекторы способны измерять минимальный сигнал 10-13 А, что соответствует 10-6 об.%. Если учитывать еще и разбавление пробы в колонке в 5 раз, то концентрация составит 5×10-6 (об. %).
Чувствительность к органическим примесям в воздухе, подающимся в детектор для поддержания горения водорода, приблизительно в 100-1000 раз ниже, чем к тем же примесям, поступающим в детектор с газом-носителем или водородом. Причина в том, что примеси в воздухе, как правило, не достигают зоны ионизации.
ПИД обладает наибольшим линейным диапазоном 106 – 107.
Устойчивость работы ПИД зависит от выбора расходов водорода, кислорода, газа-носителя. Наиболее оптимальным соотношением газ-носитель: водород: кислород является 1:1:10, причем расход водорода и газа-носителя должен быть в пределах 2-3 л/ч.
Особенности эксплуатации детектора
следить за герметичностью водородной линии не только для стабильности работы, но и для предотвращения взрыва воздушно-водородной смеси при значительной утечке водорода.
пламя водорода практически бесцветно. Если оно окрашено в желтый цвет, значит, загрязнены газовые потоки, подаваемые в пламя.
очень слабая реакция на воду и отсутствие чувствительности к неорганическим соединениям, инертным газам и водороду делают его незаменимым при анализе примесей органических веществ в воздухе промышленной зоны, сточных и природных вод.
Рисунок 2.10 – Влияние расхода водорода
Рисунок 2.11 – Влияние расхода кислорода на сигнал детектора на сигнал детектор
Присутствие воды снижает чувствительность ПИД к органическим соединениям.
Содержание воды (1,6 ± 0,6) × 10-3% меняет чувствительность ПИД на 1%. Кроме того, в присутствии воды образуются малоподвижные гидратированные ионы Н3О+*Н2О; Н3О+*2Н2О, некоторые из которых не достигают коллекторного электрода детектора.
Слабая зависимость чувствительности детектора от изменения расхода газов и температуры, а также строгая пропорциональность сигнала количеству вещества в широких пределах, делают ПИД практически лучшим детектором.
2.7.5 Электронозахватный детектор (ЭЗД)
Весьма популярный детектор вместе с ПИД и ДТП. Столь быстрое развитие ЭЗД получил в связи с необходимостью измерения малых количеств хлорсодержащих пестицидов в продуктах растительного происхождения. Он успешно используется для определения галоген-, кислород-, азотсодержащих соединений, некоторых металлорганических соединений и других веществ, содержащих атомы с явно выраженным сродством к электрону.
Система детектирования: ионизационная камера (ячейка детектора) и источник поляризующего напряжения (блок питания). Для устойчивой работы детектора необходимо обеспечить постоянную скорость образования свободных электронов в ионизационной камере, что достигается помещением в нее радиоактивного источника.
В качестве газа-носителя используют азот, аргон, гелий и другие электроно – донорные газы, способные ионизироваться под воздействием радиации с освобожденными электронами:
\[N_{2}\ \rightarrow \ N_{2}^{+} + \ e\]
Образование электронов происходит в электрическом поле между электродами детектора. Напряженность поля недостаточна для сбора всех зарядов, и начальный (фоновый) ток детектора формируется в основном электронами, подвижность которых на три порядка выше, чем подвижность ионов. Вклад ионов в ток детектора невелик, так как большая часть их рекомбинируется. При появлении в детекторе молекул анализируемых веществ, обладающих сродством к электрону, происходит захват ими свободных электронов:
\[M\ + \ e\ \rightarrow M^{-}\]
В результате, число заряженных частиц в ионизационной камере не меняется, эффективная подвижность связанных электронов резко уменьшается, и они не участвуют в процессе переноса тока между электродами. Это приводит к снижению фонового тока, который связан с количеством анализируемого компонента.
Образовавшиеся отрицательные ионы регистрируемых молекул легко рекомбинируют с ионами азота:
\[M^{-} + \ N_{2}^{+} = M + \ N_{2}\]
Это вносит дополнительный вклад в уменьшение тока детектора.
Основная трудность в достижении малого предела детектирования является большой уровень флуктуационных шумов, что неизбежно, так как для получения высокой чувствительности от проб с высокой концентрацией необходимо наличие большого количества свободных электронов.
Недопустимо присутствие в газе-носителе примесей (кислород), снижает количество электронов или их подвижность.
Уровень фонового тока обычно (1…5)×10-9, А
Уровень шума 10-13, А
Предел детектирования 5×10-10 – 10-11 мг/мл
Недостаток: узкий линейный диапазон. Очевидно, полезный сигнал изменяется, начиная с того момента, когда в детектор вводится столько вещества, что оно способно связать больше электронов, чем образуется в ионизационной камере.
Чувствительность ЭЗД резко и неравномерно увеличивается с увеличением числа атомов галогенов в молекуле, а также в ряду фтор-, хлор-, бром-, иодсодержащих соединений.
Можно сравнить пределы детектирования ЭЗД и ПИД для хлорбензолов с различным числом атомов хлора.
В то время как чувствительность ЭЗД быстро нарастает с увеличением числа атомов хлора, чувствительность ПИД лишь медленно снижается, оставаясь на довольно высоком уровне. Поэтому во многих случаях, когда для детектирования подобных смесей не нужна высокая селективность ЭЗД, удобнее применять ПИД.
Чтобы обеспечить полное участие всех частиц, расстояние между электродами должно быть довольно большим. Это вызывает увеличение объема детектора и дополнительное размывание пробы в нем. Для уменьшения инерционности иногда используют дополнительный поток газа-носителя через детектор (продувку). Продувка сохраняет эффективный объем детектора, увеличивает скорость прохождения через него анализируемого вещества и способствует достижению максимальной чувствительности без увеличения скорости газа-носителя.
Чувствительность ЭЗД изменяется с изменением температуры, поэтому детектор необходимо жестко термостатировать.
Максимальная рабочая температура определяется источником: широко используется 3Н (тритиевый), 63Ni (никелевый). Ni источник является β-излучателем со средней энергией частиц около 20 кэВ и пробегом частиц в воздухе 8 мм. Период полураспада 63Ni 125 лет. Активный слой никеля наносят на медную подложку. Максимальная рабочая температура до 3000С.
Работу ЭЗД существенно ухудшает загрязнение поверхности источника анализируемыми веществами или парами жидкой фазы, так как это уменьшает мощность излучения и меняет фоновый ток детектора. Метод может быть распространен на нейтральные вещества, если к ним предварительно химическим путем привить функциональную группу с большим сродством к электрону.
2.7.6 Термоионный детектор (ТИД)
Детектор ионизации пламени со щелочным металлом, известный под названием "термоионный", является модификацией ПИД. Наиболее высокочувствительный и селективный детектор на ФОС.
Принцип действия: увеличение ионизации солей щелочных металлов в пламени водорода при попадании в него элементорганических соединений. Процессы ионизации ТИД сосредоточены внутри пламени, тогда как в ПИД – у среза горелки.
Механизм: при введении нейтральных молекул соли щелочного металла в пламя водорода происходит их ионизация, в результате чего наблюдается резкое увеличение фонового тока (на 2-3 порядка больше ПИД). Анализируемая молекула разрушается с образованием гетероатомов, взаимодействие которых с заряженными комплексами солей щелочных металлов приводит к резкому увеличению скорости образования ионов элементорганическими соединениями. Лимитирующим процессом в таком механизме является скорость введения в водородное пламя паров солей щелочных металлов, этот процесс должен быть стабилизирован, критерием постоянства потока является фоновый ток.
Требования к стабильности газового питания детекторов, в особенности водорода, могут быть снижены до уровня ПИД.
Наибольшее отношение сигналов ТИД к ПИД наблюдается для соединений фосфора 103 – 104, при этом минимально определяемое содержание 10-5%.
Наименьшее достигаемое значение предела детектирования ТИД – 5×10-11 мг/с, типичное – (1…5)×10-9 мг/с.
Наибольшее применение он нашел в анализе пестицидов, инсектицидов и других биологически активных веществ.
2.7.7 Пламенно-фотометрический детектор (ПФД)
Селективный детектор на фосфор– и серосодержащие соединения предложен в 1966 году.
Принцип действия: измерение свечения водородного пламени при сгорании в нем фосфор– и серосодержащих соединений. В ПФД пламя обогащено водородом.
Регистрация интенсивности излучения: световой поток проходит интерференционный фильтр, который поглощает фоновое излучение пламени и далее поступает на чувствительный элемент фотоумножителя. Далее фототок направляется в электрометрический усилитель и на самописец. Выбираются две длины волны λ 526 нм (P) и 394 нм (S).
Ширина пропускания светофильтра определяет селективность и чувствительность ПФД. Применение фильтра с более узкой полосой пропускания увеличивает селективность, но уменьшает чувствительность, так как интенсивность светового потока пропорциональна квадрату ширины пропускания полосы. Использование двух светофильтров и фотоумножителей, расположенных по разные стороны от горелки, позволяет одновременно регистрировать P– и S– содержащие соединения, присутствующие в смеси.
Соотношение скоростей газов в любых режимах питания ПФД оказывает большое влияние на устойчивость его работы и зависит от конструкции.
ПФД обладает низким пределом детектирования P– и S– веществ, значение которого для лучших конструкций находится на уровне 10-9 мг/с (S) и 10-10 мг/с (P).
Для оценки факторов, влияющих на характеристики ПФД, можно пользоваться эмпирическими закономерностями, главные из которых:
чувствительность ПФД тем выше, чем выше содержание этих элементов в молекуле анализируемого вещества;
чувствительность по сере зависит от степени окисления атома серы;
присутствие фосфора в серосодержащих соединениях может искажать сигнал ПФД, аналогично, если ситуация будет наоборот;
сигнал ФОС пропорционален концентрации их в потоке газа-носителя;
сигнал ССС пропорционален логарифму потока вещества.
Характерной особенностью ПФД является зависимость чувствительности к серо– и фосфорсодержащим соединениям от присутствия в пламени других веществ.
Пример: наличие в пламени углеводородов, выходящих из колонки вместе с анализируемыми соединениями, может уменьшить или подавить пики этих компонентов.
Недостатки: гашение пламени детектора дозами элюируемого вещества, характерными для насадочных колонок. Резкое снижение максимально вводимой пробы увеличивает предел детектирования ПФД. Для его снижения применяют сложные системы выброса зоны растворителя или поддержания горения пламени.
2.7.8 Фотоионизационный детектор (ФИД)
Принцип действия: ионизация молекул элюируемых из хроматографической колонки веществ под действием вакуумного УФ-излучения и измерение возникающего ионного тока.
Процесс включает четыре стадии:
Прямая ионизация молекул вещества (АВ):
\[AB + \ h\gamma\ \rightarrow \ AB + e\]
Ионизация в результате взаимодействия АВ с возбужденными молекулами газа-носителя С:
\[C\ + \ h\gamma\ \rightarrow \ C^{*}\]
\[AB\ + \ C^{*}\ \rightarrow \ AB^{+} + \ e\ + \ C^{*}\]
Ионизация фотонами с промежуточным переходом молекул АВ в возбужденное состояние:
\[AB + \ h\gamma\ \rightarrow \ AB^{*}\]
\[AB^{*}\ \rightarrow \ AB^{+} + \ e\]
Побочные реакции, главные из которых ответственны за рекомбинацию заряженных частиц:
\[AB^{+} + \ e\ \rightarrow \ AB\]
\[C + \ e\ \rightarrow C^{-} -\]
\[AB^{+}\ + \ C–\ \rightarrow \ AB\ + \ C\]
Приведенная система не накладывает ограничений на выбор подвижной фазы хроматографической системы, поскольку потенциалы ионизации обычно используемых газов – носителей существенно выше энергии УФ – облучения.
Тем не менее, в качестве подвижной фазы лучше использовать аргон, так как по имеющимся данным, относительный отклик ФИД на равные количества регистрируемого вещества снижается в 1,5 раза при замене аргона на гелий или азот и примерно в 5 раз при переходе к СО2.
В качестве источников излучения применяются УФ – лампы аргонового, криптонового, ксенонового и водородного наполнения, испускающие фотоны с энергией 9,5; 10; 10,2; 10,9; 11,7 эВ.
Чаще используется водородная лампа, полоса с длиной волны 121,6 нм. Этот источник УФ – излучения создает наиболее интенсивный пучок фотонов, что особенно важно при определении микропримесей, а также при работе с капиллярными колонками.
Наиболее распространена водородная лампа (10,2 эВ), укомплектованная окошком из фтористого магния, выдерживающая до 225°C (длительная работа) и до 300°C и выше (в течение непродолжительных периодов эксплуатации).
Достаточно высокие потенциалы ионизации некоторых неорганических и органических соединений, превышающих энергию УФ – излучения выбранных типов ламп, исключают возможность обнаружения соответствующих молекул.
Водородная лампа не регистрирует такие соединения, как метанол, хлороформ, четыреххлористый углерод, ацетонитрил.
Не могут быть использованы в качестве растворителей проб ацетон, гексан, ароматические углеводороды, к которым ФИД обладает очень высокой чувствительностью.
С наибольшей отдачей ФИД применяется при исследовании качественного и количественного состава сложных многокомпонентных композиций нефтепродуктов и примесей токсикантов, содержащихся в объектах окружающей среды: в воздухе, почве, донных отложениях, природных и сточных водах.
Следует отметить, что серийно выпускаемые детекторы ФИД являются концентрационными, недеструктивными и практически нетребовательными к природе газа-носителя.
3 Коррекция хроматограммы
Во время работы сигнал каждого детектора хроматографа изменяется во времени.
Изменения сигнала с высокой частотой (доли секунды) называются шумом.
Изменения сигнала с низкой частотой (минуты, десятки минут) называются дрейфом.

1 – всплески, 2- шум, 3 – амплитуда, 4 – беспорядочный дрейф, 5 – дрейф, 6 – скачки
Рисунок 3.1 – Основные типы искажений нулевой линии
Шум и дрейф зависят от ряда факторов (чистота и исправное состояние детектора и его узлов, чистота и стабильность расходов питающих газов, фон колонки). Каждый детектор имеет свой набор наиболее важных параметров, критичных для достижения наилучшей чувствительности и стабильности сигнала.
В неисправном состоянии детектор может показывать повышенные фон, шум, дрейф нулевой линии. Нулевая линия может иметь "рваную" неровную структуру, сопровождаться всплесками, скачками, систематическими или случайными флуктуациями.
Для некоторых видом анализов типичен дрейф нулевой, носящий систематический характер. Он появляется при программировании температуры колонки и представляет собой нормальное явление.
3.1 Влияние системы обработки сигнала, ее механических и электронных компонентов
Всплески. Обычно обусловлены помехами в сети, плохой электроизоляцией кабелей, неисправностями системы обработки сигналов детектора. Могут вызываться включением (выключением) других приборов, а также плохими электрическими контактами в местах соединений тракта сигнала детектора. Всплески могут быть как в положительную, так и в отрицательную сторону. Всплески характеризуются малой шириной (обычно 0,2–0,4 сек).
Шум. Может являться следствием неисправности электрометрического усилителя или излучениями электронного оборудования, работающего в непосредственной близости к хроматографу.
Беспорядочный дрейф. Как правило, вызывается изменениями внешних условий – резкими изменениями температуры среды или напряжения в сети.
Скачки. Плохие контакты в местах соединений тракта прохождения сигнала.
3.2 Влияние детектора
Всплески. Положительные всплески могут быть вызваны попаданием частиц вещества в активную часть детектора. Положительными считаются всплески в том же направлении, что и хроматографические пики. Соответственно и природа возникновения сигнала детектора такая же, как и в случае регистрации детектором вещества, разделенного хроматографической колонкой. В этом случае требуется очистка детектора: продувка или промывка.
Частицы вещества могут быть внесены газами, проходящими через детектор. В том числе, для пламенных детекторов, и вспомогательными газами (водородом и воздухом). Причиной всплесков могут быть также утечки в линиях вспомогательных газов.
Для неразрушающих пробу детекторов, у которых возникновение сигнала основано на изменении концентрации вещества в рабочей камере, причиной всплесков может являться быстрые изменения расхода газа–носителя на входе или выходе детектора. Эти изменения могут быть вызваны наличием твердых частиц или жидкости в трубопроводах детектора.
Шум. Причинами высокого уровня шума могут быть утечки в газовых линиях, ненадежное присоединение колонок. В пламенных детекторах шумы могут быть обусловлены неправильным соотношением воздуха и водорода или механическим дефектом, или загрязнением сопла горелки.
Смещение нулевой линии может быть вызвано утечками в газовых линиях, неправильным выбором расходов газов, неправильными электрическими параметрами, подводимыми к активному элементу детектора.
Беспорядочный дрейф. Обычно обусловлен нестабильностью газовых потоков, температурной нестабильностью термостатов, загрязнением фильтров, загрязнением изоляции в камере пламенных детекторов.
Дрейф. Нисходящий дрейф вызван "выгоранием" химических загрязнений внутри детектора или выдуванием загрязнений из газовых линий. Восходящий дрейф нулевой линии встречается редко. Может быть вызван колебаниями Объёмных скоростей газов.
3.3 Влияние пробы
Смещение. Чрезмерное смещение обусловлено элюированием вещества – загрязнения из системы ввода или аналитической колонки. Также для аналитической колонки может быть характерен унос газом–носителем или смыв растворителем неподвижной фазы. Возможно постепенное загрязнение испарителя и колонки тяжелыми компонентами пробы. В этом случае следует отметить, что эффективность системы испаритель – колонка ухудшается. То есть, возможно изменение времени удерживания, эффективности разделения компонентов пробы, появление на хроматограмме посторонних, "ложных" пиков.
Обычно смещение зависит от температуры. Независимо изменяя температуру термостата колонок и испарителя, можно определить степень их влияния. При увеличении температуры колонок или испарителя происходит возрастание показаний детектора. При этом в некоторых случаях может оказаться достаточным кондиционирование колонки, продувка испарителя и детектора при повышенных температурах.
Беспорядочный дрейф. В изотермическом режиме вызван наличием примесей в газе–носителе или его нестабильным расходом.
Дрейф. Восходящий дрейф при программировании температуры – нормальное явление. При программировании температуры количество вещества – загрязнителя, элюируемого из колонки, не всегда возрастает пропорционально повышению температуры. В большинстве случаев при программировании температуры колонки величина дрейфа, при наличии в газе носителе вещества – загрязнителя, зависит от времени выдержки колонки на первой изотерме. Чем больше время выдержки, тем больше дрейф. В этом случае дрейф может регистрироваться довольно продолжительным пиком (несколько минут). Это лишний раз подтверждает наличие загрязнений, переносимых газом носителем.
В случае, если сигнал детектора дрейфует при повышении температуры колонки до некоторого значения и остается на этом уровне в течение второй изотермы температуры колонки, можно сделать предположение о плохом качестве хроматографической колонки. Но в реальности следует учитывать также соотношение регистрируемых концентраций компонентов пробы и величины дрейфа при программировании температуры хроматографический колонки. Абсолютная величина дрейфа (во время регистрации) должна быть ниже, чем высота регистрируемых хроматографических пиков. В этом случае она будет оказывать минимальное влияние на расчёт площадей хроматографических пиков.
Ложные пики. Обусловлены загрязнениями в системе ввода пробы или колонке выделениями из мембраны испарителя. Следует очистить узел ввода пробы и откондиционировать колонку.
В случае, если дрейф, повышенные шумы, "всплески" носят систематический характер, обусловлены особенностями конкретной методики и не являются неисправностями хроматографической системы, для их устранения можно воспользоваться соответствующими процессами коррекции хроматограммы (описаны в следующих разделах).
Для коррекции хроматограмм в "Хроматэк Аналитик" используются операции:
фильтрация;
коррекция нулевой линии;
компенсация дрейфа;
вычитание каналов.
4 Разметка пиков (интегрирование)
Интегрирование пиков (разметка) – операция вычисления параметров пиков на полученной хроматограмме. При этом пики ограничиваются базовой линией (прямой, соединяющей точки начала и конца пика на нулевой линии).
На основании полученных фигур оцениваются время удерживания, площадь и высота.
В некоторых случаях необходимо знать ширину пика, которая измеряется у его основания и совпадает с длиной базовой линии. Используется также понятие ширина пика на половине его высоты (например, для расчёта эффективности колонки).
Корректность разметки пиков оказывает большое влияние на правильность результатов количественного анализа.
В программе "Хроматэк Аналитик" использован алгоритм автоматического детектирования пиков на основе первой производной (наклона) хроматографической кривой, дополненный механизмом событий интегрирования.
Первоначально программа анализирует уровень шума хроматограммы и определяет "шум производной" – максимальный наклон касательной к кривой, которую представляют собой шумы детектора.
Далее программа проходит окном, равным заданной ширине пика по всем точкам хроматограммы, усредняет их в пределах окна и определяет производную (касательную к участку хроматограммы в границах окна).
Если производная превышает заданный порог, в средней точке окна начинается новый пик.
Следует иметь в виду, что никакой алгоритм не может в ряде случаев (сложная форма базовой линии, плохое разделение хроматографических пиков, малые пики-наездники, высокий уровень шумов и гарантировать корректную автоматическую разметку на пики, поскольку само понятие "пик" во многом субъективно и зависит от конкретно решаемой задачи. При этом правильность получаемых результатов зачастую зависит от опыта оператора. Поэтому оператор должен сообщить процедуре автоматического детектирования пиков как можно больше априорной информации, для того чтобы хроматограмма была размечена в соответствии с его представлениями.
Для капиллярной колонки характерны узкие пики, для насадочной – широкие. Поэтому для разметки пиков компонентов, разделенных на капиллярных и насадочных колонках, обычно применяются различные наборы значений параметров интегрирования.
Для достижения желаемого результата необходимо варьировать параметры интегрирования.
5 Идентификация пиков
Идентификация – отнесение пиков на хроматограмме к тому или иному компоненту из списка. При этом производится сравнение рассчитанных параметров удерживания всех обнаруженных на хроматограмме пиков с информацией, хранящейся в таблице компонентов.
Идентификация в программе "Хроматэк Аналитик 3.0" проводится по следующему алгоритму:
Составляется список всех возможных пар (пик – компонент), для пиков, попадающих в окна поиска компонентов. Таким образом, в построенном списке один пик может претендовать на несколько компонентов и наоборот.
Составленный список сортируется по удовлетворению критерия выбора пика в окне компонента. Критерии сортировки: близость по шкале времени или индекса.
Из отсортированного списка пар идентифицируются опорные пики. Компонент считается идентифицированным, если в списке пар присутствуют одноименные подтверждающие для него компоненты. При отсутствии пиков–претендентов для подтверждающих компонентов, идентификация пика не производится. После идентификации опорных пиков пересчитываются ожидаемые параметры удерживания (время, индекс) для обычных и всех прочих пиков.
Идентификация обычных пиков проводится аналогично этапу 3.
5.1 Идентификация по времени удерживания
Наиболее простой способ идентификации – сравнение времени удерживания анализируемого компонента с временем удерживания известного соединения при строго заданных условиях анализа. Для проведения идентификации пика по времени удерживания в таблице компонентов используются следующие поля:
название компонента;
время удерживания;
окно поиска по времени, %.
Окно поиска – границы области, в которой будет осуществляться поиск пика, как в положительную, так и в отрицательную сторону от заданного в таблице параметра удерживания.
На хроматограмме пик присваивается тому компоненту, заданное время удерживания которого наиболее близко к реальному времени пика.
5.2 Идентификация относительно опорных пиков
При идентификации по времени удерживания большую помощь оказывает введение опорных пиков. Например, при одновременном сдвиге по той или иной причине времен удерживания всех компонентов наличие опорных пиков поможет правильно идентифицировать вещества несмотря на то, что время их удерживания не будет попадать в окно поиска по времени.
Опорными, как правило, выбираются стоящие отдельно или большие пики. Им присваивается тип: опорный в таблице компонентов и задается увеличенное окно поиска (2–5%). В этом случае идентификация производится следующим образом:
Производится поиск опорных пиков по времени удерживания.
Рассчитывается разница между реальными и табличными временами удерживания опорных пиков и строится линейная функция сдвига времен удерживания.
Для обычных (не опорных) пиков рассчитывается ожидаемое время удерживания, исходя из заданного в таблице компонентов и функции сдвига времен удерживания.
5.3 Идентификация по индексам удерживания
Для идентификации могут использоваться относительные параметры удерживания, которые в меньшей степени зависят (в отличие от времени удерживания) от условий анализа. Одним из таких параметров является индекс удерживания – безразмерная величина, характеризующая положение пика вещества на хроматограмме относительно пиков выбранных стандартов. Если в качестве стандартов используются н-алканы, то индекс удерживания называется индексом Ковача. Выбор типа индекса (линейный или логарифмический) зависит от условий анализа. Для постоянной температуры колонки во время анализа характерна логарифмическая зависимость, при программировании – линейная. Однако между этими двумя крайними случаями нет четкой границы.
При идентификации по индексам удерживания в таблицу компонентов должны быть занесены табличные (справочные, взятые из методик) значения индексов компонентов, а также окно поиска по индексу. Необходимо опорным пикам присвоить тип "Опорный" в таблице компонентов и задать увеличенное окно поиска (2–5%) по времени.
Опорных пиков должно быть два, желательно выходящих до и после анализируемого компонента. Если в качестве опорных пиков используются н-алканы, то выбирают ближайшие по времени относительно анализируемого вещества. Если в таблице компонентов указано больше двух опорных пиков, то при идентификации выбираются те два из них (до и после компонента), которые по времени удерживания расположены ближе к интересующему веществу.
Идентификация по индексам удерживания производится следующим образом:
Производится идентификация опорных пиков по времени удерживания.
Опорным пикам присваиваются соответствующие индексы из таблицы компонентов.
Используя заданные индексы удерживания опорных пиков, программа автоматически рассчитывает индексы удерживания обычных пиков и сравнивает с табличными данными. При соответствии расчётного значения индекса ожидаемому (с учетом допустимого окна индекса), пик считается идентифицированным.
Линейный индекс удерживания:
\[l_{i} = l_{n} + \frac{(l_{n + 1} - l_{n})(t_{i} - t_{n})}{(t_{n + 1} - t_{n})}\]
Логарифмический индекс удерживания:
\[l_{i} = l_{n} + \frac{(l_{n + 1} - l_{n})(log(t_{i}^{'}) - log(t_{n}^{'}))}{(log(t_{n + 1}^{'}) - log(t_{n}^{'})}\]
Где:
Ii – индекс удерживания интересующего пика.
In, In+1 – индексы предыдущего и последующего компонентов с известной величиной индекса.
ti – время удерживания интересующего пика.
tn, tn+1 – времена удерживания пиков, соответствующие предыдущему и последующему компонентам с известными индексами.
t’ – приведенное время удерживания.
Приведенное время удерживания равно разности абсолютного времени удерживания и мертвого времени (времени нахождения неудерживаемого компонента в хроматографической системе). Мертвое время определяется экспериментально. Если мертвое время не задано, то программа принимает за него время удерживания первого пика.
Если у компонента задано более одного параметра удерживания, идентификация производится согласно приоритету шкал (время, линейный индекс, логарифмический индекс), задаваемого оператором для всей хроматограммы. Если опорные пики не заданы – идентификация всех пиков производится только по времени удерживания.
В окно ожидаемого времени или индекса удерживания одного компонента может попасть несколько пиков. В этом случае выбор пика, в зависимости от настройки, может осуществляться по следующим критериям:
ближайший к ожидаемому времени или индексу удерживания;
максимальный по высоте;
максимальный по площади.
Один и тот же пик может попасть в окна поиска разных компонентов, тогда он будет сопоставлен компоненту, имеющему меньшее время удерживания.
В общем случае, схема идентификации пиков на хроматограмме может быть следующей:
опорные компоненты – максимальные по высоте,
остальные – ближайшие по времени или индексу.
Для более точной коррекции времен и расчёта индексов удерживания рекомендуется выбирать не менее двух опорных компонентов: один в начале хроматограммы и один ближе к окончанию.
Указанная схема распознавания компонентов оказывается достаточно универсальной и гибкой для того, чтобы проблема корректного распознавания в подавляющем большинстве случаев не стояла. Все, что требуется от оператора – на этапе настройки грамотно выбрать опорные пики и в дальнейшем время от времени корректировать их ожидаемые времена удерживания по текущей хроматограмме.
5.4 Идентификация на двух каналах детекторов
Схема идентификации компонентов на двух каналах детекторов применяется для более надежной, достоверной идентификации при анализе сложных многокомпонентных проб, когда одного параметра, времени удерживания компонента далеко недостаточно.
Варианты двухканальных схем идентификации:
Последовательное детектирование. Проба проходит через разделительную колонку, попадает в первый детектор, затем без разрушения переходит во второй детектор. Необходимое условие таких схем: первый детектор не должен разрушать образец. Примеры: ДТП-ПИД, ЭЗД-ТИД, ЭЗД-ПИД.
Параллельное детектирование. Проба вводится и разделяется в хроматографической колонке. На выходе из колонки проба разделяется на два потока, каждый из которых попадает в независимый детектор. В этом случае может применяться сочетание двух любых детекторов.
Разделение с применением двух колонок различной полярности. Этот способ идентификации основан на различиях в разделительной способности двух неподвижных фаз, имеющих разную полярность. Проба попадает в устройство ввода и затем разделяется на две хроматографические колонки, имеющие разную полярность. Компоненты двигаются по колонкам с различной скоростью. На выходе каждой колонки один и тот же компонент имеет разное время удерживания. В этом варианте идентификации обычно используется сочетание двух одинаковых детекторов.
При проведении идентификации на двух каналах создаются компоненты с одинаковым именем на каждом канале детектора. При этом в столбце "идентификация" для компонента на одном из каналов устанавливается признак опорный или обычный (в зависимости от цели), на втором – подтверждающий. Идентификация с применением двух каналов может сочетаться с любыми вышеописанными приемами. Так, если для компонента не создается одноименный компонент на втором канале, его идентификация будет проведена по обычному алгоритму.
При проведении анализа с применением двух колонок различной полярности идентификация становится достаточно сложной и требует от оператора проведения правильных настроек при создании таблицы компонентов. Следующие советы позволят повысить достоверность идентификации и избежать ошибок при интерпретации хроматограмм:
Времена удерживания компонентов должны указываться максимально точно. Окна поиска должны быть небольшие с целью минимизации временных областей поиска, в которых возможно присутствие двух или более компонентов.
При идентификации на двух каналах обычный (опорный) компонент следует выбирать на том канале, где этот компонент лучше отделяется от других анализируемых компонентов и посторонних пиков. На втором канале, где пересечение областей поиска с другими компонентами более вероятно, данный компонент будет носить признак подтверждающий.
Аналогично, если сорбент колонки на одном из каналов способствует элюированию несимметричных пиков (время удерживания которых отклоняется при увеличении концентрации), компонент на этом канале должен иметь признак подтверждающий. На канале, имеющем симметрично элюируемый пик, компонент будет иметь признак обычный (или опорный).
Если различие заданного параметра удерживания двух компонентов с реальным временем удерживания пика одинаково, предпочтение в идентификации будет отдано компоненту с меньшим окном поиска. Это может быть использовано оператором, как искусственный прием, например, если присутствие одного компонента в анализируемой пробе более вероятно, чем второго. Для использования этого приема, двум близко элюируемым компонентам назначается одинаковое время удерживания и незначительно отличающиеся окна поиска (например, 3% и 2.99%).
Тем не менее, идентификация с применением двух каналов является достаточно сложным процессом и в некоторых ситуациях программа может работать некорректно.
6 Градуировка
Градуировка проводится с целью получения градуировочной зависимости, которая характеризует связь между величиной отклика детектора (площадь или высота пика) и количеством компонента в пробе. Градуировочная зависимость компонента в общем случае имеет вид:
\[Q(R) = \ K_{2}R^{2} + K_{1}R + K_{0}\]
Где:
Q(R) – количество компонента в пробе.
R – отклик детектора.
K2, K1, K0 – градуировочные коэффициенты компонента (коэффициенты чувствительности детектора к компоненту).
Для получения градуировочной зависимости проводят анализы одного или нескольких образцов с известным содержанием анализируемых компонентов.
Концентрации компонентов в градуировочных смесях в идеале должны охватывать весь диапазон измеряемых концентраций. Для получения достоверных результатов анализ каждой градуировочной смеси проводят не менее 2-х раз.
По полученным результатам строится градуировочный график зависимости отклика пика от концентрации.

Рисунок 6.1 – Градуировочный график зависимости отклика пика от концентрации
Градуировочные коэффициенты K2, K1, K0 рассчитываются исходя из градуировочного графика.
При многоточечной градуировке зависимость может быть аппроксимирована кривой любого, необязательно линейного типа. Градуировочные коэффициенты рассчитываются методом наименьших квадратов для кривой, наилучшим образом описывающей экспериментальные данные. Тип кривой для расчёта выбирается оператором.
Если градуировочная зависимость представляет собой прямую линию, проходящую через начало координат Q(R) = K1R, она может быть построена по одной точке (с применением одной градуировочной смеси с известными концентрациями компонентов).
Выбор отклика (площадь или высота) для формирования графика градуировочной зависимости не всегда однозначен. В большинстве случаев площадь является более объективным показателем, однако есть много результатов в пользу лучшей воспроизводимости высоты. Общие закономерности таковы:
Если пики узкие и высокие, то их высота измеряется более точно, чем площадь.
Для несимметричных пиков расчёты, основанные на высотах, непригодны.
Площади пиков более устойчивы к колебаниям условий анализа.
При градуировке методом абсолютной градуировки количество вещества компонента рассчитывается по формуле:
\[Q\ = \ CV\]
Где:
C – концентрация компонента.
V – объем вводимой пробы.
При градуировке методом стандарта с эффективным объемом построение графиков идет в тех же осях, однако количество вещества компонента определяется по формуле:
\[Q\ = \ \frac{CWis(\ Ris\ )}{Cis}\]
Где:
Wis(Ris) расчётное количество внутреннего стандарта.
Cis известная концентрация внутреннего стандарта в смеси.
При некоторых методах расчёта, например, методе внутренней нормализации, часто используются относительные коэффициенты отклика детектора, которые бывают известны заранее. В этом случае можно, не выполняя градуировочных измерений, выбрать функцию вида Q(R) = K1R и вручную задать значение коэффициента K1.
7 Количественный расчёт
Количественный расчёт – завершающая стадия количественного анализа, на которой производится расчёт концентраций компонентов в пробе с неизвестным содержанием анализируемых компонентов. При проведении количественного расчёта измеряют отклик анализируемого компонента и по имеющейся градуировочной зависимости рассчитывают его концентрацию.
Наиболее часто используются следующие методы количественного расчёта:
7.1 Метод процентной нормализации
Метод основан на том, что сумма площадей или высот всех пиков на хроматограмме принимается за 100%.
Этот метод не требует предварительной градуировки и предполагает ряд условий:
все компоненты анализируемой пробы элюируются из колонки;
все компоненты имеют одинаковые коэффициенты чувствительности детектора. Концентрации вычисляются по формуле:
\[Ci\%\ = \frac{\ Ri\ }{\sum_{}^{}R}\]
Где:
Ri – отклик (площадь или высота) пика.
∑R – сумма откликов всех пиков в канале хроматограммы.
7.2 Метод внутренней нормализации
В методе внутренней нормализации учитывают относительные коэффициенты чувствительности детектора к различным компонентам анализируемой смеси, заранее известные. Отклик каждого компонента пересчитывают с учетом этих коэффициентов. Сумму полученных откликов принимают за величину, равную коэффициенту нормализации. Коэффициент нормализации равен 100%, если детектируются все вещества анализируемой пробы.
В противном случае содержание недетектируемых веществ определяют другими методами и уменьшают коэффициент нормализации на величину недетектируемых компонентов. Метод внутренней нормализации не применим, если на хроматограмме присутствуют зашкаленные пики.
Концентрацию вещества вычисляют по формуле:
\[Ci\%\ = \ \frac{NM_{i}W(Ri)}{\sum_{}^{}\left( MW(R) \right)}\]
Где:
N – коэффициент нормализации.
W(R) – построенная заранее градуировочная зависимость компонента.
M – множитель.
В простейшем случае, когда градуировочная зависимость имеет вид:
\[W(R)\ = \ K1R\]
Где:
K1 – относительный коэффициент чувствительности детектора к компоненту.
Формула принимает вид:
\[Ci\%\ = \ \frac{NK1i\ Ri\ Mi}{\sum_{}^{}(KiRM)}\]
7.3 Метод внутренней нормализации для многоканальных хроматограмм
При расчёте методом внутренней нормализации иногда бывает необходимо просуммировать компоненты, детектируемые на разных каналах. Для этого нужно привести каналы к одной шкале чувствительности.
Приведение к одной шкале чувствительности производится путем расчёта поправочных коэффициентов (коэффициентов сшивки) для каждого из суммируемых каналов. Эти коэффициенты рассчитываются программой автоматически с помощью компонентов (компонентов сшивки), детектируемых одновременно на 2-х или более каналах. Формула расчёта концентрации компонента методом внутренней нормализации для многоканальных хроматограмм имеет вид:
\[Ci\%\ = \ \frac{N\ Wi(Ri)KnMi}{\sum_{}^{}\left( W(R)KnM \right)}\]
Где:
Kn – коэффициент сшивки для канала, который участвует в расчёте концентрации данного компонента.
7.4 Метод абсолютной градуировки
В методе абсолютной градуировки количество компонента в пробе определяют по предварительно полученной градуировочной зависимости. Важнейшими требованиями при проведении количественного анализа методом абсолютной градуировки являются точность дозирования образца пробы, а также строгое соблюдение условий хроматографирования при проведении градуировки и при определении содержания анализируемого компонента.
Метод абсолютной градуировки является частным случаем внешнего стандарта.
Количество вещества находится по градуировочной зависимости и корректируется с учетом множителя и общего множителя:
\[Qi\ = \ Wi(Ri)MiD\]
Концентрация компонента равна отношению количества вещества компонента к объему введенной пробы:
\[Ci\ = \ \frac{Wi(Ri)MiD}{V}\]
Где:
V – объем введенной пробы.
W(R) – построенная заранее градуировочная зависимость компонента.
M – множитель.
D – разведение.
7.5 Метод внешнего стандарта
Абсолютная градуировка является частным случаем метода внешнего стандарта.
В методе внешнего стандарта предварительно определяют градуировочную зависимость (методом абсолютной градуировки) для компонента, взятого за внешний стандарт. Для остальных компонентов используют относительные коэффициенты чувствительности детектора по отношению к внешнему стандарту, заранее известные или определенные экспериментально. Расчёт концентраций веществ методом внешнего стандарта осуществляется по формуле:
\[Ci\ = \ \frac{Ws(K1sRi)MiD}{V}\]
Где:
K1i – коэффициент чувствительности i-го компонента на данном детекторе относительно внешнего стандарта.
Ws(R) – построенная заранее градуировочная зависимость внешнего стандарта.
M – множитель.
D – разведение.
7.6 Метод внутреннего стандарта
Метод внутреннего стандарта основан на добавлении известного количества определенного вещества, называемого внутренним стандартом, к анализируемым смесям. Если относительные коэффициенты чувствительности детектора к компонентам по внутреннему стандарту не известны заранее, проводится градуировка с использованием смеси с известным содержанием анализируемого вещества и внутреннего стандарта.
Для определения градуировочной зависимости можно использовать либо метод абсолютной градуировки, либо метод внутреннего стандарта. Выбор метода расчёта градуировочных коэффициентов определяется следующими факторами:
Абсолютные коэффициенты позволяют проводить количественный расчёт, как методом абсолютной градуировки, так и методом внутреннего стандарта. Однако в этом случае необходимо дозирование точного объема пробы.
При расчёте коэффициентов методом внутреннего стандарта нет необходимости точного дозирования анализируемой пробы, а также исключается влияние изменений скорости газа-носителя и температуры колонки. Однако в этом случае может использоваться только линейная зависимость.
При количественном анализе неизвестной пробы к ней добавляют известное количество внутреннего стандарта и по полученной ранее зависимости рассчитывают содержание анализируемого компонента. Расчёт производится с учетом множителя и общего множителя:
\[Qi\ = \ \frac{QisWi(Ri)MiD}{Wis(Ris)}\]
или
\[Qi\ = \frac{\ QisWi(Ri)MiD}{Wis(Ris)}\]
Где:
W(R) – построенная заранее градуировочная зависимость компонента.
M – множитель.
D – разведение.
Обратите внимание на то, что в формуле Qis – это количество внутреннего стандарта в анализируемой пробе, и оно не пересчитывается с учетом множителя.
По градуировочной функции внутреннего стандарта вычисляется эффективный объем, который затем подставляется в формулы расчёта градуировочных функций прочих компонентов.
8 Дополнительные расчёты
8.1 Чувствительность
Чувствительность рассчитывается для каждого компонента независимо от других.
Для определения чувствительности анализа (в некоторых документах это также называется "предел детектирования") необходима хроматограмма холостого анализа. Если хроматограмма не выбрана, то расчёт данных параметров невозможен, а в отчет выводится замечание "Не выбрана хроматограмма холостого анализа. Расчёт предела детектирования невозможен", в соответствующих ячейках отчета появляется "–".
Если хотя бы для одной хроматограммы для расчёта чувствительности у компонента невозможно вычислить шум (нет участка хроматограммы), то выводится замечание "Отсутствует участок хроматограммы для определения чувствительности анализа для компонентов таких-то", а в таблице отчета в соответствующих ячейках появляется "–".
Если хотя бы в одной из хроматограмм заданная концентрация отсутствует, то в отчет выводится знак "–" и замечание "В одной из хроматограмм отсутствует заданная концентрация у {перечисление}".
Добавьте операцию в очередь обработки.
В настройках операции включите галочку – Загрузить последнюю по времени. Это позволит программе автоматически выбирать последнюю по времени хроматограмму нулевой линии.
Возможные варианты использования операции:
Вариант 1
У компонента задана функция "по справочным данным" и отсутствует стандарт.
Для каждой i-й хроматограммы j-го компонента:
\[C^{*} = \frac{2\mathrm{\Delta}xC^{'}}{h}\]
Где:
C* – чувствительность анализа, единицы концентрации те же, что и в таблице "Компоненты".
Δx – сигнал шума (размах) нулевой линии на участке холостой хроматограммы, соответствующий времени выхода компонента.
C' – заданная концентрация компонента.
h – высота пика (в тех же единицах счёта, что и \(\mathrm{\Delta}x\)).
Среднее значение чувствительности по нескольким хроматограммам:
\[\overline{C^{*}} = \frac{\sum_{i = 1}^{n}{C^{*}}_{i}}{n}\]
Где:
n – количество раз, которое встречается, компонент для расчёта в серии выбранных хроматограмм.
Вариант 2
У компонента задана функция "по справочным данным" но у компонента ЕСТЬ внешний стандарт.
Для каждой i-й хроматограммы j-го компонента чувствительность рассчитывается по формуле:
\[C^{*} = \frac{f^{станд}(2\mathrm{\Delta}xM)}{V}\]
Где:
C* – чувствительность, единицы концентрации те же, что и в таблице "Компоненты".
fстанд – функция оптимальной зависимости количества вещества от высоты пика у стандарта (при построении функции в качестве откликов используется высота компонента), если функция не построена, она вычисляется по модели для отклика "Площадь".
M – относительный коэффициент чувствительности компонента по отношению к стандарту.
Δx – сигнал шума (размах) нулевой линии на участке холостой хроматограммы, соответствующий времени выхода компонента. Если шум вычислить невозможно (нет участка хроматограммы), то замечание "Отсутствует участок хроматограммы для определения предела детектирования для компонентов таких-то", а в таблице отчета "–".
V – объем из паспорта хроматограммы (поле "Количество").
Среднее значение чувствительности по нескольким хроматограммам:
\[\overline{C^{*}} = \frac{\sum_{i = 1}^{n}{C^{*}}_{i}}{n}\]
Где:
n – количество раз, которое встречается, компонент для расчёта в серии выбранных хроматограмм.
Если функцию стандарта вычислить невозможно (отсутствуют градуировочные хроматограммы), то тогда формула для каждой i-й хроматограммы j-го компонента:
\[C^{*} = \frac{2\mathrm{\Delta}xC^{'}}{h}\]
Где:
C* – чувствительность, единицы концентрации те же, что и в таблице "Компоненты".
С' – заданная концентрация компонента.
Δx – сигнал шума (размах) нулевой линии на участке холостой хроматограммы, соответствующий времени выхода компонента.
h – высота пика в тех же единицах, что и Δx.
Вариант 3
У компонента выбрана функция в явном виде: Y=KсрХ, полином любой степени (как прямой, так и логарифмический), стандартная добавка (скорректированная функция), кусочно-линейная
(по сути, это Y=K1X + K0, но только для ограниченного диапазона).
Для каждой i-й хроматограммы j-го компонента чувствительность рассчитывается по формуле:
\[C^{*} = \frac{f(2\mathrm{\Delta}x)}{V}\]
Где:
C* – чувствительность. Единицы концентрации те же, что и в таблице "Компоненты".
f – функция оптимальной зависимости количества вещества от высоты пика (при построении функции в качестве откликов используется высота компонента). Если функция не построена, она вычисляется по модели для отклика "Площадь".
Δx – сигнал шума (размах) нулевой линии на участке холостой хроматограммы, соответствующий времени выхода компонента.
V – объем из паспорта хроматограммы (поле "Количество").
Среднее значение чувствительности по нескольким хроматограммам:
\[\overline{C^{*}} = \frac{\sum_{i = 1}^{n}{C^{*}}_{i}}{n}\]
Где:
C* – чувствительность, единицы концентрации те же, что и в таблице "Компоненты".
C' – заданная концентрация компонента.
Δx – сигнал шума (размах) нулевой линии на участке холостой хроматограммы, соответствующий времени выхода компонента.
h – высота пика в тех же единицах, что и Δx.
8.2 Проверка значимости коэффициентов
Литература:
Математика в техническом университете Выпуск XVII Горяинов Владимир Борисович, Павлов Игорь Валерианович, Цветкова Галина Михаиловна, Тескин Олег Иванович, "Математическая статистика", Москва, Издательство МГТУ им. Н.Э. Баумана, 2000 г.
Процедура может проводиться только при определении оптимальной градуировочной характеристики (ОГХ) по методу наименьших квадратов для любого типа уравнения вида Y = K0 + K1X + K2X2 + ... + KnXn кроме стандартной добавки.
После расчёта уравнения ОГХ производится проверка значимости коэффициентов при соответствующем Xi. Если коэффициент не значим, он зануляется. После зануления, снова производится пересчёт уравнения по оставшимся коэффициентам.
Варианты настройки:
Нет. В этом случае уравнение оптимальной градуировочной характеристики содержит все возможные коэффициенты согласно заданной степени полинома. Максимально возможная расчётная степень полинома на 1 меньше количества разных концентраций, используемых для определения ОГХ.
Все. Проверка производится для всех коэффицентов только 1 раз.
Граничные. Проверка производится только для коэффициента при старшей степени Kn и младшей K0. В случае незначимого Kn степень уравнения ОГХ понижается, проверка проводится снова и т. д.
Проверку проводят по критерию Стъюдента.
\[T_{i} = \frac{K_{i}}{S_{y}\sqrt{c_{(i + 1)(i + 1)}}}\]
Где:
\(K_{i}\) – коэффициент при Xi.
\(S_{y} = \frac{\sum_{i = 1}^{n}{(Y_{i} - \widehat{Y_{i}})}^{2}}{n - m}\) – остаточное среднеквадратическое: \(Y_{i}\) – фактическое значение ординаты точки на графике ОГХ, \(\widehat{Y_{i}}\) – вычисленное значение ординаты по МНК, \((n - m)\) – число степеней свободы.
\(\sqrt{c_{(i + 1)(i + 1)}}\) – диагональный элемент дисперсионной матрицы Фишера \({(F^{T}F)}^{- 1}\).
Если \(T_{i} \geq {(T}_{кр\ табл} = t_{(\alpha,n - m)})\), где \(t_{(\alpha,n - m)}\) – коэффициент Стъюдента для доверительной вероятности \(\alpha = 0,95\) и числа степеней свободы \((n - m)\), то коэффициент значим.
8.3 Расчёт средней концентрации и неопределенности
Литература:
Фаддеев М.А. "Элементарная обработка результатов эксперимента": Учебное пособие. Нижний Новгород: Изд-во Нижегородского госуниверситета, 2002.
Математика в техническом университете Выпуск XVII Горяинов Владимир Борисович, Павлов Игорь Валерианович, Цветкова Галина Михаиловна, Тескин Олег Иванович, "Математическая статистика", Москва, Издательство МГТУ им. Н.Э. Баумана, 2000 г.
Производится при расчёте нескольких хроматограмм для выдачи среднего результата. Варианты настройки:
Среднеарифметический. Классический вариант расчёта. Обычно применяется в случаях, когда погрешность результату анализа приписывается, а не рассчитывается.
Средняя концентрация и ее расчётная неопределенность среди N результатов анализа вычисляется по формулам:
\[\overline{C} = \frac{\sum_{i = 1}^{N}C_{i}}{N}\]
\[\overline{\bigtriangleup C} = \sqrt{\frac{N}{\sum_{i = 1}^{N}\frac{1}{\left( {\bigtriangleup C}_{i} \right)^{2}}}}\]
Где:
\(\overline{C}\) – среднее значение концентрации компонента.
\(C_{i}\) – значение концентрации компонента в i-й хроматограмме,
\(N\) – количество хроматограмм, в которых встречается искомый компонент.
\({\bigtriangleup C}_{i}\) – рассчитанная неопределённость концентрации компонента в i-й хроматограмме.
Если \({\bigtriangleup C}_{i}\) не определена для данного компонента хотя бы в одной из рассчитываемых хроматограмм, то расчёт неопределённости не ведётся.
Равноточный и неравноточный. Если измерения выполнены прибором одного и того же класса точности, по одной и той же методике, в одинаковых внешних условиях, одним и тем же наблюдателем (либо наблюдателями одной квалификации), то такие измерения относят к равноточным. При несоблюдении хотя бы одного из перечисленных выше условий результаты измерений классифицируют как неравноточные.
В случае равноточных измерений для хроматограмм одной пробы в паспорте в поле "Количество" должны быть одинаковые числа, в противном случае измерения считаются неравноточными.
Расчёт равноточных анализов производится в случаях, когда считается, что все повторы анализов производятся в абсолютно одинаковых условиях. При нахождении средних значений концентрации и ее неопределенности используются формулы:
\[\overline{C} = \frac{Y(\overline{R})}{V}\]
\[\overline{\bigtriangleup C} = S_{y} \times t_{(\alpha,n - m)}\sqrt{\frac{1}{N} + {\psi^{T}\left( \overline{R} \right)\left( F^{T}F \right)}^{- 1}\psi\left( \overline{R} \right)}\]
Где:
\(\overline{C}\) – среднее значение концентрации компонента.
\(Y(R)\) – уравнение зависимости количества вещества от отклика,
\(\overline{R} = \frac{\sum_{i = 1}^{N}R_{i}}{N}\) – среднее арифметическое значение откликов компонента \(R_{i}\), \(N\) – количество хроматограмм, в которых встречается искомый компонент.
\(V\) – объём пробы (значение поля "Количество" из паспорта хроматограммы).
\(S_{y} = \frac{\sum_{i = 1}^{n}{(Y_{i} - \widehat{Y_{i}})}^{2}}{n - m}\) – остаточное среднеквадратическое. \(Y_{i}\) – фактическое значение ординаты точки на графике ОГХ, \(\widehat{Y_{i}}\) – вычисленное значение ординаты по МНК, \((n - m)\) – число степеней свободы.
\(t_{(\alpha,n - m)}\) – коэффициент Стъюдента для доверительной вероятности \(\alpha = 0,95\) и числа степеней свободы \((n - m)\).
\(N\) – количество хроматограмм, в которых встречается искомый компонент.
\(\psi\left( \overline{R} \right) = \left\{ \psi 0\left( \overline{R} \right);\psi 1\left( \overline{R} \right);\psi 2\left( \overline{R} \right);... \right\}\) – матрица базисных функций для среднего отклика. Базисные функции – функции при неизвестных коэффициентах. Т. е. для уравнения \(Y(R) = \sum_{l = 0}^{4}K_{l}R^{l}\) базисные функции (при коэффициентах \(K_{l}\)): \(\psi 0 = 1\), \(\psi 1 = R\), \(\psi 2 = R^{2}\), \(\psi 3 = R^{3}\), \(\psi 4 = R^{4}\).
\(\psi^{T}\left( \overline{R} \right)\) – транспонированная матрица базисных функций для среднего отклика.
\({(F^{T}F)}^{- 1}\) – дисперсионная матрица Фишера.
Остаточное СКО:
\[\left( S_{SME} \right) = \sqrt{\frac{1}{\nu}\sum_{k = 1}^{n}{(Q_{k} - Q(R_{k}))}^{2}}\]
\(\nu = n - m\) – число степеней свободы, где \(n\) – число градуировочных точек, \(m\) – число неизвестных коэффициентов в уравнении регрессии.
\(Q_{k} = C \bullet V\) – заданное значение количества вещества, где C – концентрация, V – объём.
\(Q(R_{k})\) – вычисленное значение количества вещества по уравнению регрессии, где \(R_{k}\) – нескорректированный отклик k-й градуировочной точки.
Матрица базисных функций для уравнения вида \(C = \sum_{i = 0}^{m}{K_{i} \bullet R^{i}}\) – матрица, составленная из базисных функций \(\psi_{i}\) при соответствующих коэффициентах \(K_{i}\):
\[\psi(R) = \begin{pmatrix} \begin{matrix} \begin{matrix} 1 \\ R \\ R^{2} \\ \end{matrix} \\ R^{3} \\ \end{matrix} \\ \ldots \\ \end{pmatrix}\]
Матрица для построения дисперсионной матрицы Фишера:
\[F = \left( \begin{matrix} \begin{matrix} \frac{\psi 0(R1)}{\sigma_{1}} & \frac{\psi 1(R1)}{\sigma_{1}} & \ldots \\ \frac{\psi 0(R2)}{\sigma_{2}} & \frac{\psi 1(R2)}{\sigma_{2}} & \ldots \\ \ldots & \ldots & \ldots \\ \end{matrix} \\ \begin{matrix} \frac{\psi 0(Rn)}{\sigma_{n}} & \frac{\psi 1(Rn)}{\sigma_{n}} & \ldots \\ \end{matrix} \\ \end{matrix}\begin{matrix} \begin{matrix} \frac{\psi n(R1)}{\sigma_{1}} \\ \frac{\psi n(R2)}{\sigma_{2}} \\ \ldots \\ \end{matrix} \\ \frac{\psi n(Rn)}{\sigma_{n}} \\ \end{matrix} \right)\]
Где \(\psi_{i}\) – базисная функция.
\(Ri\)– нескорректированный отклик i-ой точки на градуировочном графике.
\(\sigma_{i}\) – погрешность концентрации (по паспорту на ГСО) i-ой точки на градуировочном графике.
Расчёт неравноточных анализов производится в случаях, когда каждый анализ считается индивидуальным (повторы производятся в неодинаковых условиях).
Для каждого индивидуального анализа:
\[C_{i} = Q(R_{i})\]
\[{\bigtriangleup C}_{i} = S_{SME} \times t_{(\alpha,n - m)}\sqrt{1 + {\psi^{T}\left( R_{i} \right) \bullet \left( F^{T}F \right)}^{- 1} \bullet \psi\left( R_{i} \right)}\]
Для нахождения средних значений концентрации и ее неопределенности используются формулы:
\[\overline{C} = \frac{\sum_{i = 1}^{N}\frac{C_{i}}{\left( {\bigtriangleup C}_{i} \right)^{2}}}{\sum_{i = 1}^{N}\frac{1}{\left( {\bigtriangleup C}_{i} \right)^{2}}}\]
\[\overline{\bigtriangleup C} = \sqrt{\frac{N}{\sum_{i = 1}^{N}\frac{1}{\left( {\bigtriangleup C}_{i} \right)^{2}}}}\]
Где:
\(\overline{C}\) – среднее значение концентрации компонента.
\(C_{i}\) – значение концентрации компонента в i-й хроматограмме.
\(N\) – количество хроматограмм, в которых встречается искомый компонент.
\({\bigtriangleup C}_{i}\) – рассчитанная неопределённость концентрации компонента в i-й хроматограмме.
Если \({\bigtriangleup C}_{i}\) не определена для данного компонента хотя бы в одной из расситываемых хроматограмм, то расчёт средней концентрации производится по формуле среднеарифметического, как описано выше.
8.4 Расчёт размаха
Обычно при количественных измерениях для повышения достоверности одну и ту же пробу хроматографируют несколько раз, и за результат анализа принимают среднее значение результатов нескольких хроматограмм. Разброс результатов количественного расчёта оценивается с помощью специального критерия, называемого Размах.
Расчёт абсолютного значения размаха осуществляется по формуле:
\[D_{i}\lbrack абс.ед.\rbrack = |C_{i\ \max} - C_{i\ \min}|\]
Где:
Сi max и Сi min – соответственно максимальное и минимальное значения концентраций вещества в одной группе анализа.
Расчёт относительного значения размаха вычисляется по формуле:
\[D_{i}\lbrack\%\rbrack = \frac{100\left| C_{i\ \max} - C_{i\ \min} \right|}{C_{i\ сред}}\]
\[C_{i\ сред} = \frac{\sum_{j = 1}^{n}C_{i}}{n}\]
Где:
Сi сред – среднее значение концентрации вещества в группе анализа.
n – количество хроматограмм в группе анализа.
8.5 Расчёт точности
Данный параметр показывает процентное отклонение расчётного значения концентрации компонента от его заранее известного значения. Расчётное значение концентрации определяется по заранее построенной градуировочной зависимости.
Таким образом, заранее известная концентрация компонента в стандартном образце сравнивается с концентрацией этого же компонента, определенной по градуировочной зависимости. Если эта точность превысит допустимое методикой анализа значение, анализ не может считаться удовлетворительным. Расчёт абсолютной точности проводится по формуле:
\[K_{i}\lbrack абс.ед\rbrack = |C_{i} - C_{i0}|\]
Расчёт относительной точности проводится по формуле:
\[K_{i}\lbrack\%\rbrack = \frac{100\left| C_{i} - C_{i0} \right|}{C_{i0}}\]
Где:
Ki – результат расчёта точности.
Ci0 – концентрация i-го в образце для контроля.
Ci – результат определения концентрации i-го в образце для контроля по градуировочной зависимости.
9 Линеаризация ПФД
В настоящее время наиболее распространенным детектором для газохроматографического анализа серосодержащих веществ является ПФД (пламенно-фотометрический детектор). Детектор отличается высокой чувствительностью, стабильностью работы и надежностью.
Однако ПФД наряду с неоспоримыми достоинствами имеет и ряд существенных недостатков, многие из которых можно обойти или минимизировать тем или иным способом.
Одним из основных недостатков ПФД является нелинейность отклика детектора от количества вещества (серы), проходящего через детектор. Эта особенность является неустранимой, т.к. имеет источником физические принципы детектирования серы данным детектором.
При избытке водорода в пламени детектора все серосодержащие летучие вещества разрушаются с образованием атомарной серы. Атомы серы вступают в реакцию друг с другом, образуя молекулу S2 в возбужденном состоянии. Возбуждение снимается испусканием квантов с характерными длинами волн, которые и регистрируются фотоумножителем детектора. В зоне пламени детектора протекают реакции:
\[S\ + \ S\ = \ S2*\]
\[S2*\ = \ S2\ + \ h\nu\]
Таким образом, мгновенная величина отклика (превышение сигнала относительно нулевого фона) детектора прямо пропорциональна концентрации молекул S2* в зоне пламени и, как следствие, пропорциональна квадрату концентрации молекул S.
Количество анализируемого вещества, прошедшего через детектор в любой момент времени есть произведение концентрации вещества в газе-носителе на его объем, прошедший через детектор за данное время.
Так как объем газа-носителя, прошедшего через детектор, пропорционален времени (объем – произведение объемного расхода на время), считается, что количество анализируемого вещества, прошедшего через детектор в малый промежуток времени (промежуток, за который изменение концентрации вещества в газе пренебрежимо мало) пропорционально произведению мгновенной концентрации вещества в газе-носителе и ширину временного интервала.
\[mi\ = \ ci\ \Delta t\]
Общее количество вещества, прошедшего через детектор есть сумма количеств вещества за малые промежутки времени на интервале от начала и до конца прохождения вещества через детектор.
\[M\ = \ \sum\ mi\]
Пик вещества на хроматограмме – двумерная геометрическая фигура в координатах время – единицы отклика детектора. Площадь пика вещества на хроматограмме есть площадь между образующей сигнала детектора и базовой линией. Площадь определяется интегрированием или последовательным суммированием прямоугольников бесконечно малой ширины и высотой, равной расстоянию от базовой линии до образующей пика, и расположенных между началом и концом пика. На практике берется ширина прямоугольников, равная 1/F (где F – частота съема сигнала детектора, обычно 25 Гц). Высота малого прямоугольника есть мгновенное значение отклика детектора и зависит от мгновенной концентрации анализируемого вещества, проходящего через детектор.

Рисунок 9.1 – Определение площади пика
\[Si\ = \ hi\ \Delta t\]
\[S\ = \ \sum\ Si\]
В случае линейной зависимости мгновенного отклика детектора от количества анализируемого вещества получается, что площадь пика есть прямая пропорциональность от общего количества вещества, прошедшего через детектор.
\[hi\ \sim\ ci\]
\[Si\ \sim\ ci\ \Delta t\ \sim\ mi\]
\[M\ \sim\ S\]
В таком случае можно во всем интервале линейности детектора использовать в качестве градуировочной зависимости формулу:
\[M\ = \ K1\ S\]
Зависимость M ~ S для линейного детектора является универсальной и не зависит ни от вещества, ни от условий проведения анализа.
Как уже говорилось ранее, детектор ПФД имеет отклик, пропорциональный квадрату мгновенной концентрации серы.
\[hi\ \sim\ ci2\]
\[Si\ \sim\ ci2\ \Delta t\]
\[S\ \sim\ \sum\ ci2\ \Delta t\]
Таким образом, имеем, что в случае квадратичности (и вообще нелинейности) отклика детектора, связь между суммарной площадью пика и количеством вещества не может быть установлена.
На практике для зависимости площади пика вещества на хроматограмме, полученной на детекторе ПФД от количества вещества используется логарифмическая зависимость.
\[lg(M)\ = \ K1\ lg(S)\ + \ K0\]
Данная зависимость с приемлемой погрешностью описывает зависимость площади пика от количества вещества в диапазоне концентраций 2 порядка.
Нами разработана и реализована в программном обеспечении математическая модель, позволяющая обойти эту особенность детектора ПФД.
Принимаем в качестве мгновенного отклика детектора некую величину (назовем его приведенный отклик) равную корню квадратному из истинного отклика.
\[h_{i}^{'} = \ \sqrt{h_{i}}\]
Получаем:
\[hi'\ \sim\ ci\]
Тогда появляется понятие приведенной площади пика, как суммы приведенных площадей малых прямоугольников
\[Si'\ = \ hi'\ \Delta t\]
\[Si'\ \sim\ ci\]
\[S'\ = \ \sum\ Si'\]
Таким образом, получаем:
\[S'\ \sim\ M\]
или
\[M\ = \ K1S'\]
Несмотря на то, что размерность приведенной площади не соответствует размерности истинной площади пика, она имеет четкий физический смысл и может быть легко вычислена по хроматограмме, что позволяет использовать линейную зависимость этого параметра от массы вещества.
Можно говорить о математической модели, позволяющей на практике "линеаризовать" отклик ПФД.
Линеаризованная зависимость сигнала ПФД от массы анализируемого вещества обладает всеми положительными чертами линейной зависимости, в том числе и универсальностью.
Это открывает путь к использованию при градуировке ПФД линейной зависимости, построенной по одной точке и проходящей через начало координат, а также методов внешнего и внутреннего стандарта.
Линеаризация по умолчанию отключена. Для ее использования задайте параметр Корректировка площадей в канале операции Поиск пиков.